Капельная ошибка
Cтраница 1
Капельная ошибка и ошибки за счет неточности при отсчетах и взвешиваниях являются общими для всех объемно-аналитических определений. Ошибка, обусловленная необходимостью введения избытка перманганата, может быть учтена на основании следующих соображений. При точных работах в результаты титрования можно ввести поправку, отняв указанный объем раствора перманганата из общего числа миллилитров его, затраченных на титрование.
[1]
Капельная ошибка определяется объемом минимальной порции реактива, добавление которой после конца титрования вызывает изменение окраски индикатора.
[2]
Таким образом, капельная ошибка уменьшается с уменьшением концентрации титрованного раствора и объема вытекающих из микробюретки капель. Другими словами, капельная ошибка пропорциональна объему капли и нормальности титрованного раствора и обратно пропорциональна количеству титруемого вещества и израсходованному на титрование объему раствора.
[3]
Чтобы уменьшить величину капельной ошибки, рекомендуется применять наконечники бюретки, оттянутые в возможно более тонкий капилляр, и понижать концентрацию титрованного раствора.
[4]
Чтобы уменьшить величину абсолютной капельной ошибки, рекомендуется применять бюретки, наконечники которых оттянуты в возможно более тонкий капилляр; при этом объем капель жидкости будет соответственно меньше. Величина относительной капельной ошибки может быть уменьшена также за счет понижения концентрации титрованного раствора.
[5]
Отдельно следует рассмотреть вопрос о капельной ошибке при тех титрованиях, при которых носик микробюретки погружен в титруемый раствор и видимых вытекающих капель нет. Это относится, например, к титрованию капиллярными микро-бюретками с ртутными затворами ( стр. В действительности при каждом движении винта из канала микробюретки выталкивается жидкость в титруемый раствор. При полном обороте винта выталкивается 0 2 — 1 мм3 раствора.
[6]
Когда титруют перманганатом калия, то капельная ошибка измеряется одной каплей раствора перманганата калия, окрашивающего титруемый раствор в светло-розовый цвет.
[7]
Также в стороне мы оставим и капельную ошибку. Об этой последней Бьеррум говорит: При титровании прибавляют титрованный раствор до тех пор, пока последнее прибавленное количество, обычно одна капля, не вызовет в растворе требуемого эффекта. Даже в тех случаях, когда этот эффект вызывается очень малой по объему каплей, например 0 001 мл реактива, все же мы можем считать, что реактив прибавлен в некотором избытке. Это — капельная ошибка, величина которой зависит от объема последней прибавленной порции титрованного раствора. Следует указать, что в некоторых случаях ( при выполнении очень точных анализов) капельную ошибку можно экспериментально определить. В анализах методами нейтрализации можно после титрования измерить концентрацию водородных ионов потенциометрическим или колориметрическим способами и по найденной величине вычислить имеющийся в растворе избыток реактива.
[8]
Эта ошибка в среднем сопоставима с капельной ошибкой микротитрования. В том случае, когда роль индикатора выполняет одно из реагирующих веществ, приходится считаться с необходимостью использования растворов более высокой концентрации, что влечет за собой некоторое увеличение капельной ощибки.
[9]
Как известно, при понижении концентрации титрованных растворов уменьшается капельная ошибка титрования.
[10]
Нас, однако, интересует не столько абсолютная величина капельной ошибки, сколько относительная.
[11]
При точно проведенном потенциометрическом титровании и правильном истолковании полученных результатов капельная ошибка и ошибка титрования могут быть значительно снижены или даже вовсе устранены. Широкому применению потенциометр и-ческих методов препятствует необходимость пользоваться электродами малых размеров, а также ряд других специфических трудностей, возникающих при исследовании очень малых объемов раствора с помощью этих методов. Следует отметить, однако, что эти трудности не являются непреодолимыми и дальнейшее усовершенствование потенциометрического титрования должно привести к разработке многих полезных методов, которые помогут значительно расширить область применения ультрамикроанализа.
[12]
Если при титровании, например, кислоты мы хотим, чтобы капельная ошибка не превышала 0 1 %, то каждая капля титрованного раствора щелочи должна нейтрализовать меньше, чем 0 001 часть первоначального количества кислоты. Если объем вытекающей из микробюретки капли равен v мл, то на титрование должно уходить не менее 1000 v мл раствора. Отсюда понятно, что желательно выпускать из микробюретки капли возможно меньшего объема. Для уменьшения объема вытекающих капель к кончику микробюретки присоединяют отрезок узкой стеклянной трубки, оттянутой в тонкий капилляр, наружную поверхность которого у отверстия смазывают тонким слоем вазелина. В зависимости от диаметра отверстия получают капли большего или меньшего объема.
[13]
Если, например, при титровании расходуется 100 капель, то капельная ошибка приближается к 1 % от определяемой концентрации, при меньшем числе капель эта ошибка возрастает. Для уменьшения этой ошибки рекомендуется первое титрование считать ориентировочным и в последующем опыте ввести на одну каплю меньше, а затем дотитровать из этой же трубки раствором, разбавленным точно в 10 раз.
[15]
Страницы:
1
2
3
4
Наиболее надежно ошибку титрования можно определить экспериментально, проводя большое число анализов стандартного образца. Это особенно необходимо при разработке нового метода анализа. Однако такая работа очень трудоемка, поэтому ошибку титрования рассчитывают из условий равновесия для каждой системы. К рассчитанным минимальным ошибкам прибавляют затем другие систематические ошибки капельную, ошибку градуировки, температурную и др. Так как этот расчет основан на применении закона действующих масс к водным растворам, в следующем разделе будут приведены некоторые конкретные примеры. [c.215]
Однонормальные растворы мало пригодны для целей титриметрического анализа как слишком концентрированные. В прибавляемой при титровании ими последней капле раствора содержалось бы, очевидно, довольно много соответствующего вещества, и поэтому так называемая капельная ошибка титрования была бы вел и ка. [c.214]
Раствор тиосульфата. Высокая чувствительность иод-крахмальной реакции обусловливает вполне отчетливое посинение -60 мл раствора от одной капли 0,01 н. раствора иода. Исходя из этого, титрованные растворы иода и тиосульфата можно готовить не 0,1 н., а 0,02 и 0,05 н. Как известно, при понижении концентрации титрованных растворов уменьшается капельная ошибка титрования. Кроме того, в данном случае имеет значение и уменьшение расхода таких сравнительно дорогостоящих реактивов, как KI и Ь. [c.401]
Так как применяемые для титрования бюретки позволяют добавлять титрующий раствор порциями не меньше одной капли, то возникает капельная ошибка титрования. Объем капли в обычных бюретках на 50 мл изменяется от 0,01 до 0,05 мл. Капельная ошибка зависит от размера капли и может быть уменьшена применением тонко оттянутых кончиков бюреток, позволяющих добавлять маленькие капли. Обычно капельная ошибка не превышает 0,01% относительных, и ею можно пренебречь. Еслн на титрование расходуется 25 мл 0,1 н. раствора, то капельная ошибка 0,01—0,03 мл, т. е. 0,04— 0,12% относительных. [c.349]
Большой практический интерес представляют методы электрометрических титрований (потенциометрических, кондуктометрических, амперометрических), для которых характерны высокая точность, чувствительность, избирательность и возможность полной автоматизации [25, 29, 46, 80, 88, 90]. Определение конечной точки титрования электрическими методами позволяет значительно снизить или вовсе устранить связанные с применением цветных индикаторов капельную ошибку и ошибку титрования. Достоинство этих методов состоит в том, что можно использовать разбавленные анализируемые и титрованные растворы. [c.18]
Ошибки титрования. При проведении титрования могут возникать следующие ошибки а) градуировки, б) температурная в) отсчета г) вытекания д) смачиваемости е) капельная. [c.115]
Содержание циркония определяют комплексометрическим или амперометрическим титрованием. Последний метод более точный, поскольку капельная ошибка меньше, а ошибка титрования, связанная с применением цветных индикаторов, отсутствует. [c.102]
При использовании же электрометрических методов определения конечной точки титрования могут быть значительно снижены как ошибка титрования, так и капельная ошибка. При этом имеет место также еще одно дополнительное преимущество— применение разбавленных анализируемых и титрованных растворов при амперометрическом, кондуктометрическом или потенциометрическом титрованиях. [c.121]
Пользование бюреткой для титрования дает еще одну ошибку, которую называют капельной ошибкой титрования. Применяемые для титрования бюретки позволяют добавлять титрующий раствор порциями не меньше одной капли. Объем капли в обычных бюретках емкостью 50 жл может изменяться в пределах от 0,01 до 0,05 мл. Величина капельной ошибки зависит от размера капли и может быть уменьшена путем применения тонко оттянутых трубок на конце бюретки, позволяющих добавлять малого размера капли. Обычно величина капельной ошибки не превышает 0,1% относительной, и ею можно пренебречь. Если на титрование рас- [c.442]
При точно проведенном потенциометрическом титровании и правильном истолковании полученных результатов капельная ошибка и ошибка титрования могут быть значительно снижены или даже вовсе устранены. Широкому применению потенциометрических методов препятствует необходимость пользоваться электродами малых размеров, а также ряд других специфических трудностей, возникающих при исследовании очень малых объемов раствора с помощью этих методов. Следует отметить, однако, что эти трудности не являются непреодолимыми и дальнейшее усовершенствование потенциометрического титрования должно привести к разработке многих полезных методов, которые помогут значительно расширить область применения ультрамикроанализа. [c.24]
Точность и воспроизводимость потенциометрического титрования с помощью дифференциального электрода несколько выше точности титрования в присутствии цветных индикаторов. Если количество титруемой кислоты не превышает 0,2 з, то систематическая ошибка в серии анализов может быть снижена до 0,3%. В случае же использования цветных индикаторов эта ошибка при работе со столь малыми количествами анализируемого вещества достигает 0,5—1%. Метод потенциометрического титрования с дифференциальным электродом позволяет работать с более разбавленными растворами. Это дает возможность достичь большой точности,, поскольку ни капельная ошибка, ни ошибка титрования не влияют на установление конечной точки при потенциометрическом методе, а все неточности обусловлены только техническими причинами. [c.143]
Когда титруют перманганатом калия, то капельная ошибка измеряется одной каплей раствора перманганата калия, окрашивающего титруемый раствор в светло-розовый цвет. Капельную ошибку можно исключить, вычтя объем капли из общего объема раствора, пошедшего на титрование. [c.349]
Мюр [981, 984] титровал горячий слабокислый раствор висмута раствором хромата или бихромата калия, устанавливая точку эквивалентности капельной пробой с нитратом серебра. Появление слаборозового окрашивания указывало на окончание титрования. Ошибка составляет 2%. Определению мешают хлориды, сульфаты, мышьяк, кальций и медь. [c.102]
V Удовлетворительные результаты дает титрование 1,5—19 мг Ре (в 50 мл раствора) раствором аскорбиновой кислоты с использованием вращающегося платинового микроэлектрода [21] при потенциале 0,0 в (относительно насыщенного каломельного электрода) в среде 0,3—1 н. соляной кислоты. Определению не мешают ионы, не проявляющие окислительных свойств, у Амперометрическое титрование с вращающимся платиновым электродом применяют для определения железа в глине, шамоте и бокситах [13]. Амперометрическое титрование Ре с ртутным капельным электродом (при потенциале 0,0 в) проводят [18, 22] на фоне 0,1 н. соляной кислоты после удаления кислорода из титруемого раствора. При определении 1—2 мг Ре ошибка составляет менее 1 %. Метод высокоселективен. Определению мешают только окислители (Си +, Ag+ и др). [c.239]
Диметилдиоксим восстанавливается [23, 217, 254] на ртутном капельном катоде ( 7, = —1,4 в). Наиболее удобно титровать при потенциале —1,85 в, так как при этом и никель, и диметилдиоксим дают диффузионные токи, и поэтому получается У-образная кривая амперометрического титрования. Для титрования можно применять натриевую соль диметилдиоксима [254]. Относительная ошибка метода менее 1%. Наилучший фон — 0,Ш ацетат натрия, при котором никель определяют в присутствии трехкратных количеств железа (III), хрома (III), алюминия. Если содержание этих элементов более высокое, их лучше отделять. [c.94]
Также в стороне мы оставим и капельную ошибку. Об этой последней Бьеррум говорит При титровании прибавляют титрованный раствор до тех пор, пока последнее прибавленное количество, обычно одна капля, не вызовет в растворе требуемого эффекта. Даже в тех случаях, когда этот эффект вызывается очень малой по объему каплей, например 0,001 мл реактива, все же мы можем считать, что реактив прибавлен в некотором избытке. Это — капельная ошибка, величина которой зависит от объема последней прибавленной порции титрованного раствора . Следует указать, что в некоторых случаях (при выполнении очень точных анализов) капельную ошибку можно экспериментально определить. В анализах методами нейтрализации можно после титрования измерить концентрацию водородных ионов потенциометрическим или колориметрическим способами и по найденной величине вычислить имеющийся в растворе избыток реактива. [c.171]
При титровании с помощью весовой бюретки последнюю наполняют, взвешивают, титруют до конечной точки и снова взвешивают. Разность между весом бюретки до и после титрования показывает вес израсходованного на титрование титрованного раствора. Определения таким способом можно производить с точностью до четвертого десятичного знака 2. Это соответствует ошибке взвешивания, так как взвешивание на аналитических весах производится с точностью до четвертого знака. Однако такое точное взвешивание практически не имеет значения, так как капельная ошибка в конце титрования значительно превосходит ошибку взвешивания. Изменение окраски в конце титрования может быть замечено в лучшем случае с точностью до одной капли , так что обычно прибавляют избыток титрованного раствора, превышающий 10 мг. Можно было бы, конечно, экспериментальным путем найти поправку на этот избыток, но это не имеет значения, так как человеческий глаз различает цветовые оттенки недостаточно точно. Поэтому взвешивание весовых бюреток с точностью, превышающей 10 мг, обычно бывает совершенно бесполезным. Если на титрование расходуется более 10 г раствора, ошибка взвешивания будет менее 0,1%. При работе с меньшими количествами, как, например, при микротитрованиях, приходится соответственно увеличивать точность взвешивания. [c.27]
Предложен метод амперометрического титрования в среде безводной уксусной кислоты с применением ртутного капельного электрода [3]. В более поздней работе применен вращающийся медный и серебряный амальгамированные электроды [4]. Конструкция электродов позволяет очень быстро получать и возобновлять слой амальгамы. Ионы нитратов титровались стандартным уксусным раствором ацетата бария в среде безводной уксусной кислоты при потенциале 1 В относительно насыщенного каломельного электрода. Максимальная относительная ошибка составляла 0,45%. Продолжительность одного определения около 10 мин. [c.72]
Если при титровании, например, кислоты мы хотим, чтобы капельная ошибка не превышала 0,1%, то каждая капля титрованного раствора щелочи должна нейтрализовать меньше, чем 0,001 часть первоначального количества кислоты. Если объем вытекающей из микробюретки капли равен v мл, то на титрование должно уходить не менее 1000 v мл раствора. Отсюда понятно, что желательно выпускать из микробюретки капли возможно меньшего объема. Для уменьшения объема вытекающих капель к кончику микробюретки присоединяют отрезок узкой стеклянной трубки, оттянутой в тонкий капилляр, наружную поверхность которого у отверстия смазывают тонким слоем вазелина. В зависимости от диаметра отверстия получают капли большего или меньшего объема. [c.141]
Между нормальностью N титрованного раствора при титровании а миллиграмм-эквивалентов некоторого вещества и объемом капли v миллилитров и капельной ошибкой Р в процентах существует зависимость выражаемая формулой [c.141]
Таким образом, капельная ошибка уменьшается с уменьшением концентрации титрованного раствора и объема вытекающих из микробюретки капель. Другими словами, капельная ошибка пропорциональна объему капли и нормальности титрованного раствора и обратно пропорциональна количеству титруемого вещества и израсходованному на титрование объему раствора. Пользуясь этими уравнениями, можно решить вопрос о необходимой концентрации титрованного раствора и о величине капли. [c.142]
Раствор KMnOi имеет яркую красно-фиолетовую окраску. В процессе титрования он обесцвечивается. Окончание титрования можно установить по появлению неисчезающей розовой окраски раствора. При этом приходится прибавить лишнюю каплю раствора KMnOj, т. е. небольшой избыток титрующего раствора. Полученная погрешность называется капельной ошибкой титрования. [c.339]
Анализируемый раствор подкисляют до pH 2—3 и на кончике шпателя добавляют твердую аскорбиновую кислоту (до полного исчезновения окраски железа) При тщательном перемешивании добавляют разбавленный раствор аммиака или буферный раствор до появления неисчезающей мути. На кончике шпателя вводят избыток КСК, разбавляют водой до 200—250 мл, добавляют 20 мл аммиачного буферного раствора с pH 10 и нагревают. Раствор светлеет и по достижении температуры 70—80 С становится светло-желтым. В теплый раствор добавляют из бюретки 0,1 М раствор комплексона III при тщателыюм перемешивании, пока раствор не станет прозрачным. Добавляют эриохром черный Т (смесь с НаС1 в соотношении 1 200) до сильной сине-зеленой окраски, и избыток комплексона III титруют 0,1 М раствором МдС12 до красной окраски и в заключение титруют раствором комплексона III до сине-зеленой окраски. Так много раз можно получить эквивалентную точку и при вычислении использовать среднее значение всех эквивалентных точек. Такой прием позволяет свести к минимуму капельную ошибку титрования и повысить точность определения. [c.79]
Второй метод — титрование фторидом — представляет собой обратный метод определения фтора при помощи солей тория (см. Фтор ). Метод основан на малой растворимости фторида тория так как ни фтор, ни торий не реагируют ни на платиновом, ни на ртутном электроде, то титрование проводят в присутствии индикатора — ионов железа (III), вводимого в раствор в виде перхлората Ре(С104)з. Фоном служит 0,2 М раствор перхлората натрия, содержащий 50% спирта (для понижения растворимости осадка фторида тория). Титруют без наложения внешнего напряжения (Нас. КЭ) с ртутным капельным электродом, но вполне можно применять и платиновый электрод, поскольку РеЗ+-ионы легко восстанавливаются и на нем. Метод позволяет определять 0,6—10 мг тория с ошибкой не выше 2%. [c.319]
Другим примером является перманганатометрическое титрование. Раствор КМПО4 имеет яркую красно-фиолетовую окраску, В процессе титрования этот раствор обесцвечивается, и конец титрования можно установить по появлению неисчезающей розовой окраски раствора, что позволяет установить эквивалентную точку. При этом приходится прибавить лишнюю каплю раствора КМПО4, т. е. небольшой избыток титрующего раствора. Полученная погрешность называется капельной ошибкой титрования. [c.423]
Поскольку ОВ потенциал описывается логарифмической зависимостью от концентрации ОВ компонентов, на кривой Е—т вблизи точки эквивалентности наблюдается скачок потенциала. Наличие достаточно большого скачка потенциала позволяет провести титрование с очень высокой точностью. Если учесть, что предел точности любого объемного титрования определяется капельной ошибкой бюретки, то минимальная ошибка титрования должна находиться приблизительно в пределах 0,1% Те. Следовательно, если ОВ титрование будет остановлено в пределах т=(0,999—1,001)те, то при этом будет достигнута максимальная точность титрования. Отсюда, помимо Ег , важное значение приобретают величины о,99эт , и A o,i /.t (скачок потенциала) [c.122]
Кроме того, в этом методе не существует ошибки, связанной с выливанием. Можно пипетировать вязкие растворы или использовать их в качестве титрантов. Так как в процессе титрования кончик капилляра опущен в титруемое вещество, а читранг дозируют винтовым механизмом, подача титранта происходит практически непрерывно, чем устраняется капельная ошибка. Отпадает трудность фиксирования положения мениска вследствие параллакса или в случае непрозрачных или окрашенных растворов. [c.116]
Титрование Мп(П) раствором перманганата калия. Во избежание больших начальных токов и выделения MnOj на электроде титрование проводят при потенциале Ц- 0,4 в [отн. меркуриодид-ного электрода (отн. МИЭ)] [594]. Были исследованы условия титрования с помош ыо ртутного капельного электрода, но получить четкую волну электролитического восстановления перманганата трудно из-за окисления ртути. Титрование лучше проводить в присутствии ионов Zn(II) [152, 594, 1098]. Определению марганца мешают ионы Fe(II), а также А1(1П), если содержание последнего превышает содержание марганца. В присутствии 3—4-кратного избытка меди титрование проводят медленно. Ошибка определения марганца в цинковых электролитах не превышает 1,3%. [c.50]
V Мышьяк (III) в концентрации больше М титруют [27] раствором РЬ(СНзСОО)4 при потенциале ртутного капельного электрода от —0,8 до —0,9 в (относительно насыщенного каломельного электрода). Еще меньшие количества As (до 3 10 молъ л) можно определять титрованием в присутствии РЬ +-ионов. Средняя ошибка определения составляет 0,7% для концентраций мышьяка 10-2-10-3 Ж- и 2,1% — для 3 10- М. [c.131]
При капельном титровании на точность определения оказывает большое влияние капельная ошибка, т. е. избыточное и неучитываемое количество титранта, находящееся в последней капле. Если, например, при титровании расходуется 100 капель, то капельная ошибка приближается к 1% от определяемой концентрации, при меньшем числе капель эта ошибка возрастает. Для уменьшения этой ошибки рекомендуется первое титрование считать ориентировочным и в последующем опыте ввести на одну каплю меньше, а затем дотитровать из этой же трубки раствором, разбавленным точно в 10 раз. Если в первом опыте затрачено 69 капель, то во втором опыте вводят только 68 капель и дотитровывают разбавленным раствором, которого понадобилось, например. [c.9]
Титрование ферроцианидом проводится на ртутном капельном электроде при потенциале —0,75 в (Нас. КЭ) в хлоридной среде (0,1 Ai) по току восстановления индия. Состав осадка отвечает формуле In4[Fe( N)6]a. Метод позволяет определять от 2 до 80 кг индия, ошибка — меньше 0,5%. Если индий присутствует вместе с другими катионами, осаждаемыми ферроциан идом, то необходимо предварительно отделить его. [c.213]
Для титрования селена по методу восстановления применяются различные восстановители, в частности аскорбиновая кислота . Титрование проводят в кислой среде (серная или соляная кислота), pH которой должен составлять 1—2. Раствор, содержащий от 5 до 30 мг селена (в виде 5еОз или SeO ) в объеме 20—80 мл, нагревают до 60° С и титруют 0,1 н. раствором аскорбиновой кислоты при —0,05 в (Нас. КЭ) с ртутным капельным электродом. 1 мл 0,1 н. раствора аскорбиновой кислоты соответствует 1,974 мг Se или 2,774 мг ЗеОг, либо, если титровали селен (VI), 1,316 мг Se или 2,116 мг SeOa. Ошибка составляет, по указанию авторов метода, около 0,3% (при pH около 2). При значениях pH более высоких, чем указанные выше, ошибка увеличивается. При титровании используется ток восстановления селена, следовательно, кривая имеет форму а. [c.292]
Эльвинг и Ольсон проводят титрование титана купфероном с ртутным капельным электродом при —0,84 в (Нас. КЭ) в 10%-иой серной кислоте. По их данным (так же как и по данным В. М. Пешковой и 3. А. Галлай), состав осадка отвечает формуле Ti( upf)4. Результаты очень точны — отклонения лежат в пределах графической ошибки при построении кривой титрования и не превышают в среднем 0,32% отн. Количество определяемого ти- [c.316]
В заключение следует упомянуть еще об одном методе, несколько отличающемся от других по своей сущностикак известно, цирконий образует в кислой среде растворимые комплексные соединения с перекисью водорода, восстанавливающиеся на ртутном капельном электроде при положительных значениях потенциала При добавлении купферона эти соединения разрушаются с образованием осадка купфероната циркония. Это дает возможность титровать цирконий в виде перекисных соединений. купфероном при -f0,4 в (МИЭ), т. е. при потенциале, отвечающем области диффузионного тока восстановления перекисного соединения купферон при этом потенциале не окисляется. Титрование ведут на фоне 2 М серной кислоты, поэтому потенциал окисления ртути сильно сдвинут в сторону положительных значений, что и дает возможность работать в указанной области потенциалов (как известно, потенциал меркур-сульфатного электрода составляет + 0,682 в). В начале титрования ток, обусловленный восстановлением перекисного соединения, несколько повышается. Вследствие образования растворимого комплексного соединения с купфероном в момент достижения отношения циркония к купферону, равного 1 1, ток достигает максимума, после чего понижается за счет образования осадка — купфероната циркония. Метод позволяет определять от 0,5 мг циркония в титруемом объеме и больше, при средней относительной ошибке 3,5%. Определению мешают Bqe ионы, осаждаемые купфероном в 2 AI серной кислоте. [c.356]
С другой стороны, малая толщина слоя жидкости при исследовании малых объемов, даже со специально разработанной техникой наблюдения, требует повышенной концентрации индикатора для возможности визуального наблюдения изменения в титруемой системе. Поэтому относительная величина капельной ошибки, связанная с объемом минимальной порции реактива, добавление которой по окончании титрования вызывает изменение окраски индикатора, в ультрамикротитровании значительно больше, чем в макротитровании добавляемое для этого количество реактива составляет значительную долю общего объема. Однако эта ошибка, связанная с присутствием большого количества индикатора, может быть учтена введением поправки на количество индикатора, а также титрованием в присутствии свидетеля. Кроме того, в объемном ультрамикроанализе для облегчения фиксации момента изменения цвета индикатора используют прием локализации его, например на частицах осадка, волокнах или в маленькой капле органической жидкости, которая не растворяется в титруемом растворе. Но и здесь появляются причины, по которым результаты титрований не всегда хорошо воспроизводятся. Так, например, при использовании приема локализации индикатора на шелковых волокнах оказывается, что интенсивность окрашивания в процессе титрования несколько уменьшается по следующим причинам. [c.112]
Во многих случаях можно экспериментально определить необходимый для перехода окраски индикатора избыток (или недостаток) титрованного раствора, а также и капельную ошибку и по этим данным вычислить исправленный титр. Когда хотят установить тптр раствора с особенно большой точностью, прибавляют его сначала из весовой бюретки, пока не останется лишь несколько капель до конца, а затем заканчивают титрование, приливая сильно разбавленный титрованный раствор из микробюретки или обыкновенной бюретки. Таким способом капельная ошибка практически устраняется. [c.44]
Наступает вполне резко, возможно, что в последний MOjMenr было прибавлено слишком много титрованного раствора. Избыток не превышает объема капли, вытекаюпхей из бюретки. Вызываемая этим обстоятельством погрешность называется капельной ошибкой. [c.141]
Так как в микрохимическом анализе пользуются разбавленными титрованными растворами (обычно 0,01 н. и слабее) и выпускают из микробюреток очень мелкие капли (около 0,01 мл и меньше), то капельная ошибка здесь незначительна. Если принять объем капли при макротитровании равным 0,03 мл, а при микротитровании 0,01 мл, то капельная ошибка, выраженная в миллиграмм-эквивалентах, соответственно равна [c.141]
Капельная ошибка при микротитровании во много раз меньше, чем при макротитровании. Определить абсолютную величину капельной ошибки довольно затруднительно (например, колориметрическим методом), во всяком случае на это было бы потрачено значительно больше времени и труда, чем на самое титрование. [c.141]
Лекция Тема: ТИТРИМЕТРИЯ
ПЛАН
1.Сущность титриметрического метода, основные понятия.
2.Классификация титриметрических методов.
3.Приготовление и стандартизация растворов титрантов. Методы пипетирования и отдельных навесок.
4.Ошибки анализа.
5.Контрольные вопросы.
1. Сущность титриметрического метода, основные понятия
Титриметрический (объемный) анализ — один из разделов количественного анализа, основанный на точном измерении объема раствора реагента (титранта), вступившего в химическую реакцию с определяемым веществом. Концентрация раствора реагента должна быть точно известна.
Раствор реагента (титранта) с точно известной концентрацией называют
стандартным, или титрованным рабочим раствором.
Наиболее важной операцией титриметрического анализа является титрование — процесс постепенного прибавления титрованного рабочего раствора к определяемому веществу. Титрование продолжают до тех пор, пока количество титранта не станет эквивалентным количеству реагирующего с ним определяемого вещества.
Момент титрования, когда титрант добавлен в количестве, эквивалентном определяемому веществу, называется точкой эквивалентности (т.э.).
Следует отметить, что на практике часто определяют не точку эквивалентности, а конечную точку титрования (к.т.т.). Эти точки часто не совпадают, но близки. Конечную точку титрования можно определять по изменению окраски раствора, по изменению физико-химических свойств титруемого раствора или же при помощи индикатора.
Чаще всего для ее определения вводят индикатор («свидетель») — вспомогательное вещество. Его действие сводится к тому, что по окончании реакции между титруемым веществом и титрантом в присутствии небольшого избытка титранта индикатор претерпевает изменения (строение или физические свойства), которые заметны визуально (меняет окраску раствора или осадка, образует осадок).
С помощью индикаторов можно установить конечную точку титрования (к.т.т.) – момент титрования, когда наблюдается изменение цвета индикатора. В идеальном случае т.э. и к.т.т. совпадают, однако в практических условиях между ними наблюдается некоторая разница. Чем больше эта разница, тем больше погрешность титрования при прочих равных условиях, поэтому следует подбирать такой индикатор, чтобы разность между т.э. и к.т.т. была минимальной.
В ряде случаев при титровании можно обходиться без индикаторов (безындикаторное титрование). Например, конечную точку титрования в перманганатометрии устанавливают в результате окрашивания, которое возникает
|
при добавлении лишней капли окрашенного раствора |
титранта KMnO4 |
|
(безындикаторный метод). |
|
|
Титриметрический метод имеет ряд достоинств: |
|
|
— высокая скорость и точность анализа, погрешность не превышает |
0,1%; |
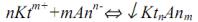
—применимость для определения различных количеств веществ;
—в одном и том же растворе часто можно определять одновременно несколько веществ;
—возможность автоматизировать титрование.
Втитриметрии применяются реакции, удовлетворяющие следующим требованиям: -реакция должна быть стехиометрична и протекать строго по заданному уравнению;
-количественно (Кр=1 ), в заданном направлении, практически до конца, с достаточно высокой скоростью и желательно при комнатной температуре; -побочные реакции и посторонние вещества не должны осложнять основную реакции;
-должна четко фиксироваться точка эквивалентности визуально с помощью индикаторов или с помощью приборов.
Правила титрования
1.Следует устанавливать титр стандартного раствора и применять один и тот же раствор в присутствии одного и того же индикатора.
2.Для титрования следует брать всегда одно и тоже количество индикатора и повторять титрование определяемого вещества несколько раз до тех пор, пока не будут получены близко сходящиеся результаты, совпадающие между собой в пределах 0,2-0,3%.
3.Необходимо брать, как правило, не более 1-2 капель индикатора, не забывая о том, что индикаторы, применяемые в методе нейтрализации, сами являются кислотами или основаниями. На их нейтрализацию также расходуется часть раствора титранта.
4.Всегда следует титровать до одного и того же оттенка окраски раствора, используя для титрования по возможности одинаковые объемы титруемого раствора.
5.Необходимо выбирать такой индикатор, который изменяет свой цвет вблизи точки эквивалентности.
2. Классификация титриметрических методов
Классификация по типу реакций:
I. Методы, основанные на реакциях, протекающих без изменения степени окисления:
а) методы кислотно-основного (перенос протонов) титрования. В их основе лежит реакция нейтрализации: Н3О+ + ОН— = 2Н2О; б) методы осадительного титрования, основанные на образовании труднорастворимых
|
веществ в процессе титрования: |
; |
|||||
|
в) |
методы |
комплексиметрического |
титрования |
основаны |
на |
реакциях |
|
комплексонообразования как с неорганическими, так и |
с органическими |
лигандами, |
например, с комплексонами (комплексометрия): 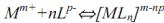 ;
;
II. Методы, основанные на реакциях окисления — восстановления (перенос электронов):
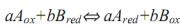 . Каждый метод получает название по стандартному раствору: перманганатометрия (титрант — раствор KMnO4);
. Каждый метод получает название по стандартному раствору: перманганатометрия (титрант — раствор KMnO4);
дихроматометрия (титрант — раствор К2Сr2О7); йодометрия (титрант раствор I2) и т.д.
Такие методы называют еше редокс-методами и их достаточно много. Классификация титриметрических методов по приемам титрования:
1.Прямое титрование. В этом случае определяемое вещество непосредственно титруют раствором реагента или наоборот.
Расчет концентрации или массы определяемого вещества рассчитывают по закону эквивалентов- вещества реагируют строго в эквивалентных соотношениях:
. Зная, что количество моль-эквивалента рассчитывается по формулам:

применим данные формулы к закону эквивалентов- вещества реагируют в объемах обратно пропорционально их нормальным концентрациям:
|
= |
, откуда |
= |
|||||||
|
2.Обратное титрование (титрование по остатку). |
К |
анализируемому |
раствору |
приливают избыток стандартного раствора (точно измеренный объем и точно известной концентрацией), избыток (остаток) не вступившего в реакцию, которого оттитровывают другим стандартным раствором (титрантом).
=
3.Способ замещения (косвенное). Используют в случаях определения неустойчивых веществ или невозможности зафиксировать точку эквивалентности при прямом титровании. К анализируемому веществу добавляют избыток специального реагента, который вступает в реакцию с анализируемым веществом. Один из продуктов взаимодействия, количество которого эквивалентно количеству определяемого вещества, оттитровывают стандартным раствором (титрантом). Расчеты производят по формулам прямого титрования.
2. Приготовление и стандартизация растворов титрантов. Методы пипетирования и
отдельных навесок
В титриметрическом методе анализа используют растворы с точно известной концентрацией — стандартными. Различают первичные и вторичные стандарты.
Первичным стандартом называют стандартным раствор, приготовленный по точной навеске. Реагент (исходное, установочное вещество) для приготовления первичного стандарта должен удовлетворять ряду требований:
1)вещество должно быть химически чистым (массовая доля примесей не выше
0,05%);
2)состав вещества должен точно соответствовать формуле;
3)вещество должно быть устойчивым при хранении;
4)вещество должно иметь по возможности большую молярную массу эквивалента, чтобы уменьшить погрешность при взвешивании.
Первичные стандарты используют как для обычных титриметрических определений, так и для установления точной концентрации растворов вторичных стандартов.
Вторичным стандартом (или установленным раствором) называют стандартный раствор, концентрация которого установлена по первичному стандарту.
Процесс определения точной концентрации вторичного стандартного раствора титрованием по первичному стандартному раствору называется стандартизацией раствора.
Способы приготовления титрованных растворов
1.Приготовление титрованного раствора по точной навеске стандартного вещества. Рассчитанную массу навески стандартного вещества для приготовления раствора заданной концентрации (первичного стандарта или титранта) взвешивают на аналитических весах на часовом стекле (или в бюксе, стакане) с точностью до
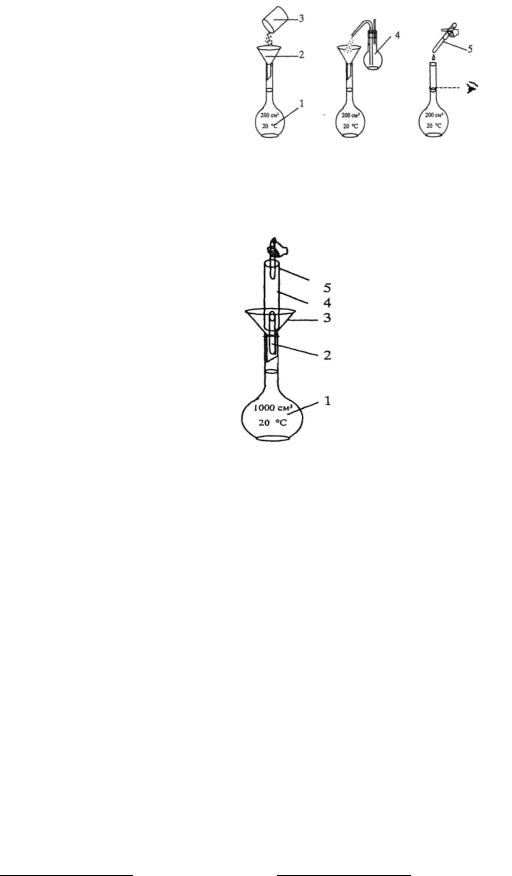
|
0,0001 г, переносят количественно (полностью) в |
||||||||||
|
чистую мерную колбу заданной вместимости, |
||||||||||
|
растворяют и1) доводят до метки дистиллированной |
||||||||||
|
водой (рис. 1). |
||||||||||
|
2. Приготовление титрованного раствора из |
||||||||||
|
фиксанала. |
||||||||||
|
Рис. 1. Приготовление стандартного |
||||||||||
|
Для |
приготовления титрантов |
и |
растворов |
|||||||
|
стандартов |
применяют |
также |
специальные |
раствора |
||||||
|
1-мерная колба; 2-воронка; 3- стаканчик или |
||||||||||
|
стандарт-титры – фиксаналы. |
||||||||||
|
бюкс с навеской; 4- промывалка с |
||||||||||
|
Фиксанал |
– раствор |
или сухое |
вещество, |
дистиллированной водой; 5- капельная |
||||||
|
пипетка |
||||||||||
|
помещенный |
в |
герметичную |
ампулу |
заводского |
||||||
|
Рис. 2. |
||||||||||
|
производства, |
содержащий |
строго |
определенное |
|||||||
|
Приготовление |
||||||||||
|
(обычно 0,1 моль-экв) количество химического |
||||||||||
|
раствора из |
||||||||||
|
соединения. |
Содержимое |
ампулы |
количественно |
фиксанала |
||||||
|
переносят |
в |
мерную колбу |
заданного объема, |
1-мерная колба; |
||||||
|
2-нижний боек; |
||||||||||
|
разбивая ампулу о вложенный в воронку боек (рис |
||||||||||
|
3- воронка; |
||||||||||
|
2). Вторым бойком разбивают верхнее углубление |
||||||||||
|
4- фиксанал; |
||||||||||
|
ампулы, с помощью промывалки через отверстие |
5- верхний боек |
|||||||||
|
тщательно промывают ампулу. Для промывки |
||||||||||
|
рекомендуется не менее, чем 6-кратный объем воды |
(по сравнению с вместимостью ампулы). Воронку также многократно ополаскивают водой и удаляют из колбы. Полученный раствор доводят до метки дистиллированной водой и перемешивают. Из фиксанала готовят как стандартные так и рабочие растворы. Это быстрый и достаточно точный способ приготовления титрованных растворов.
3. Приготовление рабочего раствора по неточной навеске (NaOH, KOH) или разбавление более концентрированного раствора (HCl, , HN ).
При этом нет необходимости рассчитывать точную навеску вещества, так как при всей тщательности взвешивания из таких веществ нельзя получить раствор точной концентрации. Поэтому для приготовления рабочих растворов навеску взвешивают на технических весах и применяют неточную мерную посуду. Для стандартизации рабочего раствора его титруют стандартным раствором. Такие растворы называют
стандартизированными или с установленным титром (вторичными стандартами).
Титриметрические определения могут быть выполнены двумя способами:
1. способом отдельных навесок, при котором берут несколько (2-3) близких по величине навесок анализируемого (или стандартного) вещества, помещают каждую в отдельную колбу для титрования, растворяют в произвольном объеме дистиллированной воды и полученные растворы титруют. Массу и процентную концентрацию вступившего вещества для каждой навески рассчитывают по закону
эквивалентов. = (г); = (%).
2. способом пипетирования – навеску анализируемого (или стандартного) вещества помещают в мерную колбу ( , растворяют в дистиллированной воде, доводят раствор до метки и тщательно перемешивают, закрыв колбу пробкой; затем отбирают пипеткой аликвотную часть ( или приготовленного раствора и титруют не менее трех раз. Если расхождение между параллелями не превышает 0,2-0,3% берут среднее арифметическое и рассчитывают по закону эквивалентов нормальную концентрацию или массу по формулам:
|
= |
∙ |
(г). |
||
Способ отдельных навесок характеризуется более высокой точностью, так как измерение объема производится только один раз по бюретке, но многократные этапы взвешивания увеличивают длительность анализа. Способ пипетирования, несмотря на несколько меньшую точность (трехкратного измерения объема, наличие этапа разбавления и т.д.) характеризуется быстротой и удобством определения.
4. Ошибки анализа
1.Ошибки взвешивания и отмеривания. Применяемые в анализе аналитические весы и мерные приборы обычно имеют погрешности. Например, аналитические весы обычно
имеют точность до 4-го знака после запятой, мерные приборы (пипетки, бюретки) – до 0,02- 0,03 см3. Для повышения точности на аналитических весах не рекомендуется брать навеску
меньше 0,1000 г. При работе на бюретках объемы отмериваемых растворов должны быть не менее 20 см3.
2.К ошибкам измерения относится также капельная ошибка. При титровании из бюретки минимальная порция раствора равна 1 капле. Объем капли обычно равен 0,02-0,03 см3,
поэтому при титровании возникает так называемая капельная ошибка. Для ее уменьшения необходимо расходовать на титрование 20-25 см3 титранта, оттягивать кончик бюретки для уменьшения объема капли.
3.Химические ошибки вызваны различными химическими причинами. Например, индикаторы точки эквивалентности могут изменять свою окраску не в этой точке. Это вызывает появление индикаторной ошибки, так как раствор определяемого вещества перетитровывается или недотитровывается титрантом. Для устранения индикаторной ошибки необходимо применять индикаторы, дающие переход окраски в области, близкой к точке эквивалентности. При титровании слабых кислот и оснований возникают ошибки, вызываемые малой ионизацией кислоты или основания и неполным протеканием кислотно-основной реакции. Для уменьшения ошибок титрование проводят не менее трех раз и используют так называемый контрольный опыт, который осуществляют в одинаковых условиях (реагенты, индикатор), но без определяемого вещества. Объем титранта, израсходованный на титрование контрольной пробы до изменения окраски индикатора, потом вычитают из результатов анализа. Уменьшения ошибок можно добиться, аккуратно выполняя процесс титрования. Последние порции титранта добавляют по каплям, чтобы не перетитровывать анализируемый раствор.
4.Систематические и случайные ошибки.
Систематические ошибки возникают постоянно из-за неправильности аналитических весов, мерной посуды, аналитических приборов. Для устранения систематических ошибок необходимы проверка весов, калибровка мерной посуды и других измерителей.
Случайные ошибки возникают вследствие различных причин: добавлена лишняя капля титранта, неточно взята навеска вещества. Устраняют случайные ошибки, проводя не менее трех параллельных анализов. Средний результат трех, параллельных определений в большей или меньшей мере свободен от случайных ошибок.
5. Абсолютные и относительные ошибки. Абсолютные ошибки выражают в абсолютных цифрах, относительные – в процентах от результата анализа.
5. Контрольные вопросы
1.Сущность титриметрического метода анализа. Классификация титриметрических методов (с примерами).
2.Перечислите требования к титриметрическим реакциям.
3.Дайте определения следующим способам выражения концентрации растворов: титр, титр по определяемому веществу, нормальность, молярность, массовая доля вещества в растворе. Их размерность. Как можно перейти от одной концентрации к другой (приведите формулы)?
4.Дайте определение химического эквивалента и фактора эквивалентности вещества.
5.Что такое стандартный (рабочий, титрованный) раствор.
6.Понятие первичного стандарта и вторичного, привести примеры.
7.Что такое установочные вещества и какие требования к ним предъявляются?
8.Какие рабочие растворы называются приготовленными, а какие – установленными?
9.Какие существуют способы титрования? Дайте определение каждому способу, объясните их сущность и приведите примеры.
10.Перечислите основные правила титрования.
11.Какие могут иметь место ошибки в титриметрическом методе анализа.
12.В чем отличия между точкой эквивалентности (т.э.) и конечной точкой титрования
(к.т.т.)?
13.Опишите технику основных способов приготовления титрованных растворов. 14. В чем сущность способов пипетирования и отдельных навесок?
Лекция Тема: МЕТОД КИСЛОТНО-ОСНОВНОГО ТИТРОВАНИЯ
(НЕЙТРАЛИЗАЦИИ, ПРОТОЛИТОМЕТРИИ) ПЛАН
1.Сущность кислотно-основного титрования, классификация метода.
2.Индикаторы метода нейтрализации, теории индикаторов.
3.Применение метода нейтрализации.
4.Контрольные вопросы.
1. Сущность кислотно-основного титрования, классификация метода
Кислотно-основное титрование – это метод определения кислот, оснований, солей, основанный на реакции взаимодействия между протолитами (протолитометрия)
– кислотой НА и основанием В: НА + В = А— + НВ+
В водных растворах – это реакция нейтрализации: Н3О+ + ОН— = 2Н2О.
Поэтому метод кислотно-основного титрования также называют методом
нейтрализации.
Реакция кислотно-основного взаимодействия:
—протекает быстро,
—практически необратима,
—протекает строго стехиометрично,
т.е. удовлетворяет требованиям, предъявляемым к реакциям, применяемым в титриметрическом анализе.
Титрантами метода являются растворы сильных кислот и оснований: HCl, H2SO4, NaOH, KOH. Эти вещества не соответствуют требованиям, предъявляемым к

стандартным веществам, поэтому концентрацию титрантов устанавливают стандартизацией их растворов.
Взависимости от природы титранта метод нейтрализации подразделяется на:
—ацидометрическое титрование (от латинского слова «acid») — титрант кислота –
НС1 или Н2SO4), определяют основания и соли, гидролизующиеся по аниону. В качестве первичных стандартов чаще используют гексагидрат тетрабората натрия Na2B4O7·10H2O, безводный карбонат натрия Na2CO3;
—алкалиметрическое титрование (от латинского слова «alkaly») -титрант щелочь –
NaOH или Ba(OH)2), определяют кислоты и соли, гидролизующиеся по катиону. Первичным стандартом выступаютдигидрат щавелевой кислоты H2C2O4·2H2O,
2. Индикаторы метода нейтрализации, теории индикаторов
Растворы кислот и оснований, как правило, бесцветны, и реакции между ними не сопровождаются какими-то заметными внешними эффектами. Точка эквивалентности
вкислотно-основном титровании определяется по изменению рН раствора. Поэтому для определения характера среды и для приблизительной оценки рН используют кислотно-основные индикаторы – органические красители, структура и окраска которых зависит от величины водородного показателя рН.
Молекулярная и ионная формы индикаторов имеют различную окраску. Кроме того, изменение окраски связано с таутомерией молекул индикатора.
Существуют одноцветные индикаторы, бесцветные в кислой среде и окрашенные
вщелочной (например, фенолфталеин), и двухцветные (например, метиловый оранжевый), характеризующиеся различной окраской в кислых и щелочных растворах (табл. 1).
Таблица 1. Некоторые кислотно-основные индикаторы
|
Индикатор |
рН |
РТ |
Изменение окраски |
Окраска в точке экви- |
|
валентности |
||||
|
Метиловый фиолетовый |
0-1,8 |
1,2 |
Желтая-фиолетовая |
Зеленая |
|
Тимоловый синий |
1,2-2,8 |
2,0 |
Красная-желтая |
Оранжевая |
|
Метиловый оранжевый |
3,1-4,4 |
4,0 |
Красная-желтая |
Оранжевая |
|
Бромкрезоловый зеленый |
3,9-5,4 |
4,5 |
Желтая-синяя |
Зеленая |
|
Метиловый красный |
4,4-6,2 |
5,5 |
Красная-желтая |
Оранжевая |
|
Бромтимоловый синий |
6,0-7,6 |
7,0 |
Желтая-синяя |
Зеленая |
|
Тимоловый синий |
8,0-9,6 |
9,2 |
Желтая -красная |
Оранжевая |
|
Фенолфталеин |
8,2-9,8 |
9,0 |
Бесцветная-красная |
Розовая |
|
Тимолфталеин |
9,3- |
9,6 |
Бесцветная-синяя |
Голубая |
|
10,5 |
Каждый индикатор характеризуется интервалом перехода окраски ( рН) — интервалом значений рН, внутри которого индикатор изменяет окраску, за его пределами преобладает одна из форм индикатора.
Рассмотрим равновесие в растворе кислотного индикатора:
|
HInd (бесцветная форма) ↔ |
+ In (окрашенная форма) |
||
|
= |
выразим концентрацию : |
||

Установлено, что человеческий глаз способен замечать резкое изменение цвета в растворе если соотношение концентраций молекулярной (окрашенной) формы и ионной
(бесцветной) формы будет равно:
т.к. отрицательный логарифм константы ионизации – это силовой показатель
|
–lg K= рК , |
рН= рКlg |
= рК+1; рН= рКlg |
= рК-1 , то соответственно |
||
интервал перехода окраски (ΔрН) рассчитывают по формуле:
ΔpH = pKi ± 1,
где Ki — константа ионизации индикатора.
Интервал перехода окраски зависит от природы индикатора и его свойств. Чем меньше интервал перехода окраски, тем ценнее индикатор. Значение рН, при котором заканчивается титрование в присутствии данного индикатора, называется показателем титрования (рТ). Изменение окраски происходит, как правило, при равных значениях концентраций молекулярной и ионной форм индикатора, поэтому во многих системах рТ= pKi.
Требования к кислотно-основным индикаторам:
резко различная окраска индикатора при близких значениях рН;
минимальный интервал изменения окраски;
контрастный переход окраски;
стабильность окраски индикатора;
обратимость изменения окраски.
Теории индикаторов
Существуют различные теории индикаторов, каждая из которых по своему объясняет поведение кислотно-основных индикаторов в кислых и щелочных средах.
Ионная теория индикаторов (Оствальда 1894 г). В связи с тем, что кислотно-
основные индикаторы представляют собой слабые кислоты или слабые основания, любой такого рода индикатор диссоциирует в растворе согласно уравнению:
|
слабая кислота (донор протонов) |
HInd ↔ |
+ In |
|
слабое основание (акцептор протонов) |
IndОН ↔ In |
+ |
Окраска раствора, в котором индикатор находится в молекулярной форме, отличается от окраски раствора, в котором индикатор находится в ионной форме.
Так, молекулы кислотного индикатора фенолфталеина HInd бесцветны, а его анионы In окрашены в малиновый цвет: достаточно к раствору, содержащему фенолфталеин, прибавить 1—2 капли щелочи, как введенные -ионы станут связывать катионы с образованием слабого электролита — молекул воды. При этом равновесие диссоциации индикатора сместится вправо, и накопление анионов In вызовет окрашивание раствора в малиновый цвет.
И наоборот, если к раствору фенолфталеина прилить несколько капель кислоты, то повышение концентрации одноименного иона Н+ будет подавлять диссоциацию молекул индикатора. Равновесие сместится влево, и раствор обесцветится.
Аналогичным образом объясняют поведение лакмуса, молекулы которого окрашены в красный цвет, а анионы — в синий; нейтральные растворы лакмуса имеют промежуточную фиолетовую окраску. Это подтверждает, что цвет водного раствора индикатора зависит от соотношения между его молекулярной и ионной формами.
Таким образом, переход одной окраски, присущей молекулярной форме кислотноосновного индикатора, в другую, свойственную его ионной форме, происходит под влиянием
или -ионов, то есть зависит от рН раствора.
Хромофорная теория индикаторов. Поведение индикаторов, объясняемое ионной теорией индикаторов, не объясняла изменение окраски индикатора при долгом хранении, а также обратимость окраски индикатора, эти явления объясняла хромофорная теория индикаторов, согласно которой изменение окраски индикаторов связано с изменением структуры их
|
молекул, внутримолекулярной перегруппировкой, вызываемой действием |
или |
-ионов. |
|
Согласно хромофорной теории, цветность органических |
соединений, |
|
обусловливается присутствием в |
них хромофоров «носителей цвета» группировок |
|
атомов с сопряженными связями, |
обеспечивающие поглощение видимого света из-за |
|
сравнительно легкого возбуждения электронов π-связи, например: —N=N—, —N , |
|
—NO, =C=C=, =C=O. |
|
|
Присутствие других групп атомов ауксохромов, например, |
, —N , —NHR, |
—самих по себе не придающих окраску органическому соединению, но влияющих на свойство хромофоров, углубляя цвет окрашенного вещества.
По хромофорной теории в процессе изменения рН раствора меняется строение молекул кислотно-основных индикаторов. Это явление обусловливается бензоидно-
хиноидной таутомерией.
При изменении рН среды раствора или при диссоциации хромофоры могут перегруппировываться. Перемена окраски у индикаторов является результатом изменений в их внутреннем строении. У одноцветных индикаторов окраска изменяется в связи с появлением или исчезновением хромофоров. У двухцветных индикаторов эти изменения обусловлены превращением одних хромофоров в другие.
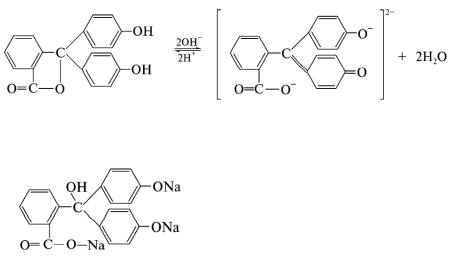
Типичным одноцветным индикатором является фенолфталеин. Молекула фенолфталеина имеет три бензольных кольца и одно из них может претерпевать хиноидную перегруппировку, которая является хромофором, а также молекула содержит две гидроксильные и одну карбоксильную и они тоже изменяются. При рН < 8 его молекулы не содержат хиноидной группировки и поэтому бесцветны. Однако при добавлении раствора щелочи к раствору фенолфталеина (рН=8) происходит перегруппировка атомов в молекуле (депротонизация) с образованием хиноидной группировки, которая обусловливает появление малиновой окраски раствора:
Дальнейшее увеличение рН до 13—14 вызывает некую перегруппировку, в результате чего получается трехзамещенная соль, лишенная хиноидной группировки и поэтому бесцветная:
Вследствие этого фенолфталеин обесцвечивается при действии большого избытка щелочи, например натрия гидроксида.
3. Применение метода нейтрализации
Методом нейтрализации в клинико-биохимических лабораториях, а также в контрольно-аналитических аптечных лабораториях определяют общую кислотность желудочного содержимого, т. е. свободную и связанную соляную кислоту, органические кислоты и кислореагирующие фосфаты, титруя свежий исследуемый материал 0,1 н. раствором едкого натра. Метод нейтрализации используется и для
определения титруемой кислотности и щелочности мочи, величины pH кишечного сока, крови.
В пищевой промышленности метод нейтрализации находит самое широкое применение. При помощи этого метода определяют карбонатную жесткость воды, кислотность молочных продуктов, хлебобулочных и кондитерских изделий, квашеной капусты, безалкогольных напитков и др.
Под жесткостью понимают суммарное содержание в воде солей кальция и магния. Общую жесткость определяют комплексонометрическим методом, а карбонатную или временную жесткость — методом нейтрализации. Различают воду мягкую (общая жесткость до 2 ммоль-экв/дм3), средней жесткости (2-10 ммольэкв/дм3) и жесткую (более 10 ммоль-экв/дм3).
Карбонатная жесткость (временная) зависит от содержания в воде гидрокарбонатов кальция и магния. Она почти полностью устраняется кипячением, при котором гидрокарбонаты разлагаются:
|
Са(НСО3)2 = CaCO3+ СО2 + |
2О |
|||
|
Mg(НСО3)2 = MgCO3+ СО2 + |
2О |
|||
|
Жесткость принято |
выражать |
в ммоль-экв/дм3 и рассчитывается по формуле: |
||
|
Ж |
V (HCl) С(1/1HCl) |
1000 . |
||
|
V (Н2О) |
||||
Постоянную жесткость, которая не устраняется кипячением определяют обратным титрованием: к пробе воды добавляют избыток стандартного раствора карбоната натрия N и упаривают досуха, затем сухой остаток растворяют в дистиллированной воде и титруют стандартным раствором соляной кислоты:
|
Ж= |
∙1000. |
||
|
О |
|||
Определение кислотности хлеба.
Кислотность хлеба обусловливается наличием в нем, главным образом, молочной и уксусной кислот, которые образуются при брожении теста. Умеренная кислотность способствует лучшему усвоению хлеба и придает ему приятный вкус. Большая кислотность хлеба вредна, так как может повысить процессы брожения в органах пищеварения.
За один градус кислотности хлеба принимают 1 мл c(l/1 NaOH) = 1 моль/л, израсходованного на нейтрализацию кислот в 100 г хлеба.
Кислотность ржаного хлеба не должна превышать 12°, а пшеничного -70. Расчет кислотности:
|
Пересчет на 1,0 н раствор NaOH: |
Vпересчет |
V (NaOH) * c(NaOH) |
||||||||
|
1.0 |
||||||||||
|
Объем 1,0 н NaOH необходимый на титрование 250 мл вытяжки: V250 |
Vпересчет * 250 |
|||||||||
|
Va |
||||||||||
|
На 100 г продукта потребуется: |
||||||||||
|
кислотность |
V250 |
*100 |
. |
|||||||
|
m |
||||||||||
Определение кислотности продуктов кондитерского производства.
В кондитерском производстве кислотность принято выражать в условных единицах
(градусах). Под градусом кислотности понимают объем (см3) раствора гидроксида натрия с
концентрацией равной С(l/1 NaOH) = 1 моль/л, необходимого для нейтрализации кислот и кислых солей, содержащихся в 100 г анализируемого изделия.
Кислотность продуктов кондитерского производства (карамель, мармелад, пастила, шоколад, халва, печенье) устанавливают титрованием водной вытяжки (экстракта).
Кислотность кондитерского изделия (град) рассчитывают по формуле:
Аналитическая химия —
Количественный анализ
| Индекс материала |
|---|
| Нормальность растворов. Грамм-эквивалент |
| Страница 2 |
| Страница 3 |
| Страница 4 |
| Страница 5 |
| Все страницы |
Страница 4 из 5
Однонормальные растворы мало пригодны для целей титриметрического анализа как слишком концентрированные. В прибавляемой при титровании ими последней капле раствора содержалось бы, очевидно, довольно много соответствующего вещества, и поэтому так называемая капельная ошибка титрования была бы велика.
Это относится и к вдвое более разбавленным — полунормальным раствор.ам (0,5 н.). Гораздо чаще пользуются в десять раз более разбавленными — децинормальными (0,1 н.) или даже в 50 раз более разбавленными — двусантинормальными (0,02 н.) растворами. Первые содержат 0,1, а вторые 0,02 г-экв соответствующего вещества в 1 л (или такое же количество миллиграмм-эквивалентов в 1 мл).
Удобство пользования точно 0,1 н. или точно 0,02 н. и т. д. растворами заключается в том, что при одинаковой нормальности растворов реакции идут между равными объемами их. Например, на титрование 25,00 мл 0,1 н. раствора любой щелочи пойдет как раз такой же объем 0,1 н. раствора любой кислоты и т. д.
Причину этого понять нетрудно. В 1 мл 0,1 н. раствора любого вещества содержится 0,1 мг-экв, а в 25 мл 0,1-25 = 2,5 мг-экв. Поскольку при титровании на реакцию затрачивается всегда одинаковое количество миллиграмм-эквивалентов обоих реагирующих веществ, она должна протекать между равными объемами 0,1 н. растворов их. Это справедливо и во всех других случаях, когда для реакции берут растворы одинаковой нормальности.
Если нормальность растворов различна, то раствора, нормальность которого больше, пойдет при титровании в соответствующее число раз меньше по объему. Например, на нейтрализацию 20 мл 0,1 н. раствора кислоты расходуется 20 мл 0,1 н., или 10 мл 0,2 н., или 5 мл 0,4 н. раствора щелочи и т. д. Следовательно, затрачиваемые при титровании объемы растворов обратно пропорциональны их нормальностям. Если объем и нормальность одного из растворов, реагирующих между собой при титровании, обозначить соответственно через V1 и Ni, а другого через V% и Л/г, то можно написать:
| < Предыдущая | Следующая > |
|---|
Ошибки титриметрического анализа также подразделяют на систематические и случайные. Обычно систематические ошибки очень невелики. Причины их будут рассмотрены далее при описании отдельных методов здесь же ограничимся только одним примером. [c.165]
Опшбки в титриметрическом анализе в зависимости от их происхождения могут быть подразделены на методические ошибки, которые связаны с особенностями метода титрования, и специфические ошибки, связанные с особенностями данного метода. [c.349]
Присутствие в растворе белковых веществ и коллоидов, а также нейтральных солей обычно тоже влияет на интервал перехода индикаторов и хотя для титрования применяют лишь те индикаторы, у которых так называемые белковая и солевая ошибки невелики, все же при высоких концентрациях белковых веществ или солей в растворах эти ошибки могут стать значительными. Чтобы исключить влияние всех указанных выше факторов на окончательный результат анализа, каждый раз, когда приходится вести титрование при нагревании или в присутствии неэлектролитов, большого количества солей и т. д., следует устанавливать титр рабочего раствора в тех же самых условиях. Это правило является вообще одним из основных в титриметрическом анализе. [c.253]
Различают прямое титрование, основанное на непосредственном взаимодействии анализируемого вещества и титранта, и обратное титрование, в котором процессу титрования предшествует вспомогательная реакция. Последний метод характеризуется несколько более высокой ошибкой, так как количество измерений при его выполнении возрастает. Для уменьшения суммарной ошибки анализа необходимо, чтобы объем раствора титранта (при выбранной навеске анализируемого вещества) был возможно большим, а ошибка в определении концентрации этого раствора— возможно меньшей. Обычно относительная средняя квадратичная ошибка результатов анализа титриметрическим методом составляет 0,1—0,5%. [c.342]
Ошибки титриметрического анализа подразделяют на систематические и случайные. Обычно систематические ошибки очень невелики. [c.317]
Типичной и наиболее широко распространенной методической погрешностью титриметрических методов анализа является индикаторная ошибка. Она возникает при фиксировании конечной точки титрования. [c.64]
Однонормальные растворы мало пригодны для целей титриметрического анализа как слишком концентрированные. В прибавляемой при титровании ими последней капле раствора содержалось бы, очевидно, довольно много соответствующего вещества, и поэтому так называемая капельная ошибка титрования была бы вел и ка. [c.214]
В титриметрическом анализе часто встречается методическая ошибка, связанная с добавлением избытка реагента по отношению к теоретическому количеству, необходимому для изменения окраски индикатора, по которой судят о конце реакции. В конечном итоге правильность всего анализа определяется спецификой того самого явления, которое лежит в основе определения. [c.60]
Ошибки титриметрического анализа могут быть вызваны следующими причинами. [c.322]
Как будет показано позже, при рассмотрении титрования с внешними индикаторами ошибку, связанную с отбором проб, можно сделать исчезающе малой. Метод равного помутнения, предложенный в 1832 г. Гей-Люссаком, явился одним из первых методов титриметрического анализа. Впоследствии он был нспользован для весьма точного определения атомных весов галогенов и серебра. [c.320]
Каждому методу анализа присущи свои ошибки, которые могут отсутствовать в других методах. Например, ошибки, связанные с потерей вещества при прокаливании, наблюдаются в гравиметрическом анализе, но их нет в титриметрическом анализе. Ошибки, связанные с применением индикаторов, характерны для титриметрического анализа, но отсутствуют в гравиметрическом анализе. Указание на эти ошибки дано при каждом отдельном методе. Есть ошибки, которые характерны для всех методов количественного анализа. Наиример, взвешивая на аналитических весах, можно всегда сделать ошибку, равную 0,0002 г. В тщательно проводимом анализе неорганических веществ относительная ошибка не должна превышать 0,1%. Поэтому навеска вещества для анализа не должна быть меньше 0,2 г. [c.283]
В титриметрическом анализе получаются ошибки при отсчетах на градуированной посуде (бюретках). Например, пользуясь бюреткой на 50 мл, при каждом отсчете мы делаем ошибку 0,02 мл. При отсчете объема в 10 мл ошибка не должна превышать 0,2%. Поэтому надо в случае бюреток на 50 мл работать с объемами растворов не менее 20 мл. [c.283]
Ошибки методические. Эти ошибки зависят от особенностей применяемого метода анализа, например от не вполне количественного протекания реакции, на которой основано определение, от частичной растворимости осадка, от соосаждения вместе с ним различных посторонних примесей, от частичного разложения или улетучивания осадка при прокаливании, от гигроскопичности прокаленного осадка, от течения наряду с основной реакцией каких-либо побочных реакций, искажающих результаты титриметрических определений, от свойств примененного при титровании индикатора и т. д. Методические ошибки составляют наиболее серьезную причину искажения результатов количественных определений, устранить их трудно. [c.48]
Каждому методу анализа свойственны свои специфические ошибки. Например, в гравиметрическом анализе имеют место ошибки, связанные с потерей вещества при промывании и прокаливании осадков. В титриметрическом анализе — ошибки, связанные с применением индикаторов. Наряду с этим имеются ошибки, свойственные всем или многим методам количественного анализа, [c.303]
Операции титриметрического анализа также выполняются с некоторыми, хотя и сравнительно небольшими, ошибками. Считают, что всякое титриметрическое определение включает в себя 1) ошибку определения титра раствора 2) ошибку титрования анализируемого вещества. Первая зависит от точности взвешивания стандартного вещества и правильности измерения объема раствора. Вторгся определяется точностью титрования, т.е. правильностью установления точки эквивалентности с помощью индикатора. Например, если капля раствора, прибавляемая к исследуемой жидкости, имеет слишком большой объем, то этим создается вероятность добавить не строго эквивалентное, а большее количество вещества. [c.165]
Кондуктометрическое титрование расширяет область применения титриметрического анализа, так как благодаря ему становится возможным титрование окрашенных и мутных растворов, когда переход окраски индикатора трудно наблюдать визуально более -точно устанавливается конечная точка при титровании слабых кислот и оснований при кондуктометрическом титровании можно использовать многие реакции осаждения и комплексообразования при анализе смеси веществ повышается точность определений. Относительная ошибка определения находится в пределах 0,1—2% в зависимости от определяемых концентраций. [c.93]
Ошибки в отсчетах по бюретке являются главным источником ошибок в титриметрическом анализе. Особенно часто подобные ошибки допускают начинающие химики, занимая неправильное положение, при отсчете (рис. 92). Относительная ошибка отсчета, вместо допустимого значения 0,1%, может достигнуть 0,3% ли даже 0,6%. [c.109]
Индикаторная ошибка. Своеобразным видом методической ошибки титриметрических методов анализа является индикаторная ошибка. Эта ошибка возникает в связи с тем, что индикатор вступает в реакцию взаимодействия с титрантом либо несколько раньше, либо несколько позже точки эквивалентности. Поскольку взаимодействие определяемого компонента и индикаторного вещества -с титрантом подчиняется законам химического равновесия, момент вступления индикатора в реакцию определяется как прочностью обоих образующихся соединений (константой образования), так и соотношением концентраций искомого компонента и индикатора. Из всех этих величин только концентрация индикатора не является закрепленной и может варьироваться в тех или иных пределах. [c.33]
Другим примером постоянной ошибки служит избыточный объем реагента, необходимый для изменения окраски в титриметрическом анализе. Объем этот обычно мал и не зависит от общего объема реагента, затраченного на титрование. И снова относительная ошибка будет тем выше, чем больше общий объем. Очевидно, одним из путей снижения постоянной ошибки будет выбор разумного количества пробы в соответствии, конечно, с методом анализа. [c.61]
Достоинства потенциометрического метода анализа. Метод потенциометрического титрования имеет при прочих равных условиях ряд достоинств по сравнению с методами визуального титриметрического анализа он более чувствителен, при его использовании исключается субъективная ошибка определения точки эквивалентности. Метод дает возможность проводить определения в мутных и окрашенных растворах и даже в вязких пастах, где невозможно использование индикаторов, и дифференцированно титровать не анализируемые другими методами многокомпонентные смеси веществ без предварительного разделения. [c.43]
Преимущества потенциометрического метода титрования. Потенциометрическое титрование при прочих равных условиях имеет ряд преимуществ по сравнению с визуальными титриметрическими методами анализа. Метод потенциометрического титрования более чувствителен, при использовании его исключается субъективная ошибка, возникающая при визуальном нахождении момента завершения химической реакции, т. е. конечной точки титрования. Этот метод дает возможность определять вещества в мутных и сильно окрашенных растворах, дифференцированно (раздельно) титровать компоненты смесн веществ в одной и той же порции раствора и, наконец, автоматизировать процесс титрования, так как измеряемой величиной является электрический параметр. [c.37]
При определении содержания добавочных компонентов допустима большая ошибка определения [а = 2. .. 5. ..10% (отн.)], особенно при определении небольших содержаний (<10″ %). Вследствие таких требований к точности определения основных и добавочных компонентов для определения первых применяют преимущественно химические методы анализа, для вторых — физико-химические методы. Из химических методов большое применение, благодаря их быстроте, находят титриметрические методы с различными способами определения точки эквивалентности. При особо высоких требованиях к точности прибегают к гравиметрическим методам анализа. Среди физико-химических методов определения добавочных компонентов особенно широкое применение нашли электрохимические методы анализа (полярография, кулонометрия) и оптические (фотометрия). При определении не очень малых количеств элементов (>1%) применяют также различные варианты объемных методов анализа. [c.399]
Систематические ошибки. Систематические ошибки обусловливаются многими причинами. К ним относятся, например, ошибки, зависящие от особенностей метода анализа. Используемые конкретные реакции данного метода могут протекать не вполне количественно, что связано с обратимостью химических процессов. Вместе с осадком могут соосаждаться и посторонние римеси, увеличивая массу осадка. Осадки даже наименее растворимые имеют какую-то частичную растворимость, что уменьшает массу осадка. Осадки могут частично разлагаться при прокаливании, впитывать водяные пары или поглощать газы из атмосферы. В растворе могут происходить побочные реакции. В титриметрических методах некоторые ошибки связаны с используемым индикатором и т. п. К этому типу ошибок относятся ошибки, связанные с личными качествами самого аналитика. Например, не все способны точно уловить момент перемены окраски раствора в процессе титрования, не всегда точно улавливаются мелкие деления на шкале приборов. Иногда [c.215]
В вычислении результатов титриметрических определений наименее точная цифра — число миллилитров титрующего раствора, израсходованного на титрование. Поскольку сотые доли миллилитра отмечаются лишь приблизительно, можно принять, что максимальная ошибка отмеривания не менее 0,02 мл. Ошибка от натекания также равна 0,02 мл. Таким образом, общая ошибка может доходить до 0,04 мл . При общем расходе титрующего раствора 20 мл это составит 0,2% отн. Отсюда следует, что, беря для анализа 1 г, вполне можно проводить отвешивание с точностью до 1 мг это дает относительную ошибку в 0,5 мг, или 0,05%. Если на титрование расходуется меньше 20 мл [c.11]
Ошибки эталонов и стандартов. Определение содержания отдельных компонентов во многих методах химического анализа опосредовано через применение разного рода стандартов и эталонов. Таковы методы фотометрического, эмиссионного спектрального, атомно-абсорбционного, газохроматографического анализов, полярографические, амперометрические, кон-дуктометрические, радиохимические и многие другие методы. В последнее время в титриметрических методах получили [c.35]
Преимущества метода кулонометрического титрования также ощутимы в тех случаях, когда для проведения анализа требуется малое количество реагента. Регулируя силу тока, можно легко и с высокой точностью вводить в раствор небольшие порции реагента, тогда как в классических титриметрических методах дозирование малых объемов даже сильно разбавленных растворов приводит к значительным ошибкам. [c.43]
Систематические ошибки. Систематические ошибки обусловливаются многими причинами. К ним относятся, например, ошибки, зависящие от особенностей метода анализа. Используемые конкретные реакции данного метода могут протекать не вполне количественно, что связано с обратимостью химических процессов. Вместе с осадком могут соосаждаться и посторонние примеси, увеличивая массу осадка. Осадки даже наименее растворимые имеют какую-то частичную растворимость, что уменьшает массу осадка. Осадки могут частично разлагаться при прокаливании, могут впитывать водяные пары или поглощать газы из атмосферы. В растворе могут происходить побочные реакции. В титриметрических методах ошибки могут быть связаны с используемым индикатором и т. п. К этому типу ошибок относятся ошибки, связанные с личными качествами самого аналитика. Например, не все могут точно уловить момент перемены окраски раствора в процессе титрования, не всегда точно улавливаются мелкие деления на шкале приборов. Иногда учащиеся ориентируются на определенные данные или на цифры, которые получают работающие рядом, а не на свои собственные. Это так называемые психологические ошибки, зависящие от настроения самого исполнителя. [c.227]
Малые количества углекислоты (несколько ч. на млн.) определить очень трудно, так как в воздухе содержится значительное количество СО2. Поэтому химический анализ связан с трудностью определения поправки холостого опыта. Вследствие того, что аммиак и С02 образуют химическое соединение, обычные методы газового анализа «здесь оказываются неприемлемыми. В работе [443] предложен титриметрический метод определения СО2 в безводном аммиаке, основанный па связывании углекислоты в виде карбамата аммония, отделении аммиака отгонкой при комнатной температуре, обработке остатка разбавленной серной кислотой, поглощении выделяющейся СО2 гидроокисью бария и титровании избытка последнего щавелевой кислотой. Этим методом при содержании углекислоты 0—50 ч. на млн. абсолютная ошибка равна 1,5 ч. на млн. [c.257]
На результат анализа могут оказывать влияние и случайные ошибки. Чтобы устранить их, титриметрическое определение повторяют несколько раз и берут среднее из них. Однако, вычисляя средний результат, допускают отклонения не более 0,3%. Результаты определений, отличающиеся на большую величину, отбрасывают при вычислении среднего арифметического. Например, если в четырех определениях нормальности раствора едкого натра были получены величины 0,1134, 0,1135, 0,1142 и 0,1136, то число 0,1142 отбрасывают, оно отличается от наименьшего числа 0,1134 на 0,8%. Из остальных величин берут среднее арифметическое. [c.318]
Весьма важное значение имеет правильное вычисление результатов титриметрического анализа. Все вычисления рекомендуется выполнять со всей тщательностью и внимательно, так как правильные результаты титрования из-за ошибок в расчетах дают неверный результат. Всякое определение включает два рода ошибок ошибку в концентрации титрующего раствора и ошибки титрования определяемого вещества. Эти ошибки компенсируются в том случае, если концентрация титруюшего раствора установлена в тех же условиях, что и титрование анализируемого образца. Влияние случайных ошибок можно устранить, повторяя титрование несколько раз. Отклонение от среднего результата не должно превышать 0,3% относительных. Поэтому отсчеты объемов по бюретке необходимо вести с точностью до 0,02— 0,03 мл. Например, три последовательных титрования дают 25,06 25,03 25,03 мл. Средний результат титрования должен быть записан в виде 25,04 мл. Если отклонения превышают допустимую величину, то такие результаты титрования не должны приниматься во внимание при вычислении среднего результата. Для повышения точности измерения объема применяют бюретки малого диаметра или весовые бюретки. [c.351]
Проведенный выше раэбор систематических ошибок хими-t e Koro аяализа не претендует на исчерпывающую полноту. Из рассмотрения исключены некоторые виды ошибок, например, ошибка натекания и капельная ошибка в титриметрических методах анализа. Некоторые виды систематических ошибок только упомянуты. Основное внимание и наибольшее количество примеров посвящено ошибкам традиционных методов гравиметрического, титриметрического и фотометрического анализов. Такой стиль изложения оправдан целью данного раздела—дать общее представление о систематических ошибках химического анализа, способах их обнаружения и оценки и методах их уменьшения. Детальный разбор всех известных источников ошибок должен входить как составная часть в теорию и практику каждого отдельного метода химического анализа, ибо каждому методу присущи свои специфические ошибки». Удачным примером в этом плане может служить руководство по (фотоколориметрическим и спектрофотометрическим методам анализа М. И. Булатова и И. П. Калин-кина (Л, Химия , 1976, 376 с.), где этому вопросу уделено большое внимание. Однако сказанное в равной мере относится и к любым другим химическим и физическим методам, [c.48]
В качестве титруюш его раствора при аналогичном определении может быть использован метапериодат [1176]. В работе [59] проведено титриметрическое определение ртути нри осаждении ее перйодатом калия в виде Hg5(100)2, растворении осадка в кислоте и иодометрическим титровании. При определении от 0,2 до 0,02 г Hg(II) ошибка равна +0,2%. Для анализа соединений Hg(II) можно также использовать данный метод, предварительно восстановив Hg(II) до Hg(I). [c.90]
Элементный бром в водах определяют титрованием раствором соли Мора по N,N-диэтил- г-фeнилeндиaминy [739, 741] согласно описанию на с. 76. Для его определения в водных растворах рекомендованы экстракционные методы с фотометрическим и титриметрическим окончанием [157]. Они требуют небольшого расхода времени, но ошибки анализа достигают 20—50%. Для качественного определения брома можно применять реакции, рассмотренные в главе III. [c.179]
Для анализа производственных вод применяется титриметрический метод определения сульфидов и цианидов потенциометрическим титрованием, разработанный в ВУХИНе [ 1 ]. Данный метод значительно сокращает время анализа, количество реактивов. Время определения составляет 10—15 мин, ошибка — 1 — 2 % (отн.) (химические методы требуют [c.59]
К ошибкам этого же типа в значительной мере можно отнести также индикаторные ошибки в титриметрических методах, поскольку во многих случаях их можно предрассчитать. В какой-то мере возможна приближенная оценка отдельных видов методической ошибки и в других методах химического анализа. Однако обычно методические ошибки включают в себя отдельные слагаемые, обусловленные не только неполнотой протекания равновесных процессов или параллельным протеканием сопроцессов , но и неравновесностью отдельных стадий химико-аналитических процессов, т. е. связанные с кинетическими факторами. [c.25]
При титровании следовых количеств хлорид-ионов возможна ошибка за счет реакции НС1 с СО,. Такую ошибку можно корректировать введением поправки [720]. Ошибку титрования увеличивает скоагулировавшийся осадок. Поэтому для предотвращения коагуляции к концентрированным растворам хлоридов лития,, натрия, калия, магния, кальция добавляют 5—10 мл 0,1%-ного раствора агар-агара. В качестве титранта используют 0,01— 0,1 N AgNOg (чаще всего 0,05А ). Метод Мора применяют в основном для анализа вод [633, 771]. В настоящее время он все больше вытесняется другими титриметрическими методами, исключающими использование серебра. Ограничивает также применимость этого метода необходимость проведения анализа в нейтральной срвде, что исключает присутствие в ана.иизируемых объектах тяжелых металлов. Поэтому гораздо чаще применяют метод Фоль-гарда, позволяющий проводить титрование в кислой среде. [c.36]
Большинство методов количественного ультрамикрохимического анализа основано на измерении какого-либо параметра, функционально связанного с массой. Это, прежде, всего, титриметрические и фотометрические методы. Важным преимуществом физико-химических методов является то, что при их использовании нет необходимости брать навески на высоко точных и чувствительных ультрамикровесах, так как микронавеска может быть взята и отбором аликвотной части раствора. При анализе жидкостей измеряют объем, взятый для анализа. В физических методах, как, например, эмиссионном спектральном, рентгеноспектральном, масс-спектральном анализе навески нЗ микровесах вообще не берут. Конечно, и в этих методах точность анализа зависит во многом от погрешности приборов. Во всяком случае в ультрамикроанализе, так же как и в микроанализе, случайные ошибки приборов составляют, по крайней мере, треть погрешности, вызываемой химическими факторами [c.183]
Анализ сплавов никеля. Предложено несколько методов, в зависимости от определяемых количеств вольфрама. Титриметрический метод [746] применяют для определения 0,5—5% W из навески 0,1 г сначала восстанавливают W(VI) до W(III) свинцом, затем вводят избыток Fe(III), восстановленное железо оттитровывают К2СГ2О7. Для определения 0,04—5% W разработан [153] экстракционно-фотометрический метод с помощью роданида и хлорида N,N N»-тpифeнилгyaнидиния. При определении 0,05 — 1,0% W и 1—5% W ошибка определения составляет соответственно + 10% и + 2—5%. Флуориметрический метод [561] позволяет определять 0,5—3% W из навески 0,05—0,1 г. [c.178]
Для определения сантимиллиграммовых количеств азота в органических соединениях предложен титриметрический метод, состоящий в разложении вещества серной кислотой в запаянной кварцевой трубке при нагревании, пропускании реакционной смеси через ионообменную смолу основного характера и титровании образовавшегося NH4OH раствором HGIO4 с потенциометрической регистрацией КТТ [782]. Продолжительность анализа 12 мин. Определяемый минимум 30 мкг N. Абсолютная ошибка +0,25%. Металлы мешают определению. [c.182]