Разложение по Кьельдалю для определения азота в породах и силикатных минералах производилось при помощи концентрированной серной кислоты в запаянных пробирках. В этом случае температура разложения может быть значительно выше, чем при обычном методе. Время полного выделения азота зависит от температуры разложения. При анализе силикатных минералов и изверженных пород минимальная продолжительность разложения составляет 90 мин при температуре 420° для осадочных пород продолжительность разложения может быть уменьшена до 60 мин. Метод является быстрым и точным. Его точность значительно выше, чем при разложении в обычных приборах Кьельдаля, так как в последнем случае ошибки могут возникать в результате толчков при кипении, загрязнения аммиаком из воздуха и минеральных веществ, оседающих на стенках колбы, а также в некоторых случаях, например при анализе слюды, в результате прилипания минерала к стенкам колбы выше поверхности серной кислоты и вследствие этого неполного разложения пробы. [c.161]
Диэтнлтолуамид, как известно, служит активной основой репеллентных жировых и эмульсионных кремов, лосьонов. Для опре,деления диэтилтолуамида в кремах ДЭТА используется. метод Кьельдаля. Анализ длится около 3 суток, относительная ошибка определения составляет 10%. [c.105]
Этим методом количество белка в продукте определяют значительно быстрее, чем методом Кьельдаля, он доступен для любой лаборатории. Метод проверен для зерновых культур. Относительная ошибка его составляет в среднем 2—5%. Кроме того, этот метод применяют и при исследовании продуктов животного происхождения. Дальнейшая разработка его для определения белка в корнеплодах и клубнях имеет большое практическое значение. [c.56]
Важным является также вопрос о вариабельности данных по содержанию белка и аминокислот в пищевых продуктах. На основании материалов, поступивших из отраслевых подкомиссий МВК нами были обобщены данные по вариабельности содержания белков и аминокислот в пищевых продуктах [12]. Вариабельность содержания белка зависит от природы продукта. В животных продуктах коэффициент вариации (относительное среднеквадратичное отклонение) равен 5—10%, для растительных (зерно, бобовые, фрукты) 15—20%. При этом внут-рилабораторная методическая ошибка сходимости (внутрилаборатор-ный коэффициент вариации) при определении азота по Кьельдалю, как отмечалось выше, не превышает 1 %. Межлабораторная воспроизводимость (межлабораторный коэффициент вариации) не превышает 2— [c.287]
Еще один тип методических ошибок иллюстрируется рис. 4-1. Определение азота в органических соединениях по Кьельдалю основано на окислении образца концентрированной серной кислотой азот обычно превращается в сульфат аммония. Соединения, содержащие пиридиновое кольцо, например никотиновая кислота, могут не разрушиться полностью в условиях анализа. Отрицательная ошибка в результатах аналитиков 3 и 4 связана, вероятнее всего, с неполным окислением. [c.60]
Метод Кьельдаля иногда обесценивается еще и тем, что при его применении не соблюдают некоторых из мер предосторожности, необходимых для получения точных результатов. В последнее время рядом исследователей [61—63] были пересмотрены условия, необходимые для того, чтобы весь азот при разложении любого белка полностью превращался в аммиак. Имеется ряд удовлетворительных методов измерения количества аммиака, образовавшегося при разложении. При надлежащем внимании к технике аналитического определения эти методы не содержат источника значительной ошибки (см. статью III). [c.17]
Косвенное определение, основанное на измерении объема азота. Это определение проводится обычным методом Кьельдаля (см. ХП1. б). Правда, при этом в присутствии содержащих азот примесей (например, биурета, циануровой кислоты или солей аммония) возможны ошибки. [c.220]
В работе [20] описана модификация микроопределения азота в ароматических нитро- и полинитросоединениях методом Кьельдаля, включающая их предварительное восстановление. В [172] микрометод Кьельдаля усовершенствован применительно к анализу полимерных соединений для разложения анализируемого вещества используют не концентрированную H2SO4, а дымящую H2SO4 (в смеси с K2SO4 и селеном), что позволяет сократить продолжительность разложения полимерных образцов. Образующийся NHg определяют фотометрически (без перегонки) с реактивом Несслера. Ошибка определения не превышает 0,3%. [c.182]
Определение азота является скорее проблемой элементарного анализа, чем проблемой анализа белков, а применение микрометода Кьельдаля почти универсально. Чибнел с сотрудниками [175] показали, однако, что в настоящее время желание ускорить анализ привело к заниженным и переменным результатам и необходимо большее время разложения, чем указывается в современных методах. Очевидно, что ошибка определения азота проявится в любых аналитических данных по аминокислотам. Однако, если проводить работу тщательно, определение азота более точно, чем любое определение аминокислот, [c.61]
Прямое определение по Несслеру продуктов разложения, полученных по Кьельдалю, пожалуй, применяется более широко, чем метод обычной перегонки [89—93]. Образующаяся окраска измеряется при помощи разнообразных фотометров и колориметров. Эта методика экономит некоторое время, и она также более целесообразна для клинических целей, вследствие широкого применения колориметров при клинических анализах. Метод является менее точным, чем перегонка, но не исключено, что вводимые ошибки не больше, чем ошибки, присущие другим этапам аналитического метода. Образование аминов [61, 76] при разложении, несомненно, вводит ошибки при работе по этому методу. Шере [94] произвел сравнительное изучение методов перегонки и Несслера и пришел к выводу, что последний достаточно точен для клинических целей. Интересное изменение, которое устраняет большую часть критики метода Несслера, состоит в применении гипобромита для окисления аммиака в субстрате [95, 96]. Избыток гипобромита определяется иодометрически. Применение фосфорномолибденовой кислоты [96] при разложении, повидимому, нежелательно [15]. [c.24]
При работе с изотопом газообразный азот для анализов обычно приготовляется, как и в методе Кьельдаля, воздействием на аммониевую соль раствора гипобромита ш елочных металлов. Перед смешиванием оба реагента должны быть освобождены от растворенного воздуха. Смешивание следует проводить под вакуумом [153, 155]. Выделившийся азот с помощью теплеровского насоса перекачивается в напускной баллон. При определении кислорода последний обычно переводится в углекислый газ. В тех случаях, когда он присутствует в виде воды, последнюю взбалтывают с известным количеством обыкновенного углекислого газа, вследствие чего последний принимает равновесную концентрацию 018 016 характерную для всей системы [156]. Вода затем вымораживается, а Og откачивается для анализа. Изотоп С обычно анализируется в форме СОд, получаемого в качестве продукта сгорания или при мокром окислении. Углекислый газ вымораживается погружением в сосуд Дьюара с жидким воздухом, чтобы можно было откачать кис,лород, после чего Og переводится в баллон масс-спектрометра [157]. Углекислый газ должен быть сухим и, кроме того, не должен содержать водорода, поскольку присутствие ионов Н+, образующихся при диссоциации последнего, ведет ко вторичным реакциям с образованием ионов HGOj [158]. Этот эффект является функцией давления в ионном источнике и может привести к ощутимым ошибкам при вычислении содержания С из отношения массы 45 к массе 44. Анализ на содержание стабильных изотопов серы обычно производится с SOg. [c.100]
НОЙ желез и цервикального гликонротеина вычислили значения от 16,1 до 17,5%. Необходимо вводить большие поправки на азот, содержащийся в других компонентах гликопротеинов, таких, как гексозамины и сиаловые кислоты, и ошибки в определении этих веществ будут понижать точность рассчитанных значений для белка. Общее содержание азота легче всего определять по методу Кьельдаля. Автор нашел, что очень точные результаты дает методика Чибнелла и сотр. [125]. Время разложения можно уменьшить с 8 до 2 час без снижения точности, если заменить сульфат меди в катализирующей смеси на то же (по весу) количество сульфата ртути [118]. При этом необходимо ввести 5% (вес/объем) тиосульфата натрия в 30%-ный (вес/объем) раствор едкого натра, прибавляемый для нейтрализации серной кислоты перед отгонкой аммиака. [c.152]
Следует, однако, принять во внимание, что оценка степени очистки вируса по отношению к общему азоту или белку не всегда достоверна. Так, при опредеяении азота по методу Кьельдаля гетероциклические азотсодержащие вещества трудно минерализуются. Ошибка при определении азота увеличится если на одном из этапов очистки применяли высаливание сульфатом аммония даже с последующим диализом препарата. Метод определения белка по Лоури 513] также имеет свои недостатки, так как для построения калибровочного графика должен быть использован белок данного вируса, а не какой-либо другой. [c.149]
Ход анализа, который я вам описала взят из ИНСТРУКЦИИ ПОЛЬЗОВАТЕЛЯ НА ДИСТИЛЛЯЦИЮ Velp UDK pade 20 of 25 » Метод Кьельдаля для определения протеина в сое» АОАС, «Официальные методы анализа» , метод 979.09 (920.87) Конкретно на анализ зерна сои ушло при титровании отгона 31,21мл 0,1н серной кислоты.
Это неправильный ход анализа, не правильный как с практической (удобство в работе и прочее), так и с теоретической (рекомендации АОАС) стороны.
Фаза сжигания 30 минут при 150 градусах это выброшенное время — там ничего не происходит. АОАС (или уж руководство по эксплуатации) рекомендует сразу ставить на 60 минут при 420 градусах. Согласитесь, это довольно большая разница.
Следующий момент — 30 мм кислоты на титрование это очень много. Если у вас холостой опыт в пределах 1-1.5 мл, то для титрования образца я рекомендовал бы использовать 15 мл кислоты. Это даст вам уверенность в точности получаемого результата и довольно значительную экономию времени и денег. Меньше образца брать — сгорать будет быстрее, меньше кислоты лить, меньше катализатора, меньше щелочи на нейтрализацию, меньше кислоты на титрование — все это будет экономить ваши силы и деньги лаборатории.
По своему опыту я рекомендую брать навеску 500-600 мг образца для зерна пшеницы или ячменя, а там белка 10-16%, вы же берете 1 грамм сои, где белка 40%. Прибор отлично позволяет определить данные количества белка/азота, и не надо его перегружать лишним образцом. Тогда он не будет ползти вверх до самых небес и спокойно будет сгорать. Единственное, на что необходимо обращать внимание — точность титрования, если вы титруете руками с помощью стеклянной бюретки или у вас очень грязная (по азоту) вода, дающая 2-3 мл холостого опыта — сильно снижать объем навески не рекомендуется (МНОЙ! ЛИЧНО МНОЙ! Будт спрашивать — так и скажите! ![]() ).
).
Поэтому ставьте печке программу 420 градусов СРАЗУ. Минут на 40. Навеску — 400 мг ну плюс-минус (для сои). Постарайтесь ее хорошо размолоть и тщательно гомогенизировать. Если параллельные не будут сходиться — просто уделите дополнительное внимание гомогенизации образца после размола. Катализатора можно брать одну таблетку (если вы делаете его сами — 3.5-4 грамма). Если гореть будет плохо — попробуйте другие катализаторы, медный катализатор не самый хороший, селен, окись ртути (как в ГОСТе), окись титана… все средства хороши. Кислоты 7 мл для сжигания достаточно.
Если за 40 минут при установке в горячий блок сжигания образцы сгорать не будут — увеличьте чуть-чуть время или уменьшите массу навески.
А ИНСТРУКЦИЮ ПОЛЬЗОВАТЕЛЯ НА ДИСТИЛЛЯЦИЮ Velp UDK верните тому, кто ее вам дал. Перевод не мой — я не отвечаю за дурь других. Не хотите говорить кто так учит работать — ваше право, но я вам честно говорю — вас учат неправильно.
Анализ белка по Кьельдалю. Коэффициент пересчета
В последнее время коэффициенту (фактору) пересчета общего аммонийного азота, определённого по методу Кьельдалю в массовую долю белка стало уделяться много внимания. Связано это как со вполне понятным стремлением установить точное содержание белка в объекте, так и с абсолютным непонятным желанием сделать это административным путем.
Проблема вариаций коэффициента пересчёта и в частности от содержания небелкового азота (далее НБА) существует давно, и никто этого не скрывает. Эта проблема обсуждается, имеются большое количество опубликованных литературных данных по теме, а также значительное количество частных мнений конкретных лиц и лабораторий, которые используют в своей деятельности анализ белка по Кьельдалю. В данной методике существуют ряд других проблем (кроме собственно коэффициента пересчета) здесь и зависимость результата от состава белковой фракции пробы, и от способа обработки пробы, и от реологии и состава самого объекта. Понятно, что для разных типов образцов и соответственно для различного вида деятельности лаборатории эти проблемы могут либо присутствовать в различной степени, либо вообще отсутствовать. Давайте сосредоточимся на самой близкой нам теме, на необработанном сыром молоке и на небелковом азоте именно в нём.
Предыстория.
Собственно, о методе и его истории написано много, мы тоже писали здесь. Так что разумно перейти сразу к сути осуждаемого вопроса. Откуда взялся коэффициент и сколько должно быть небелкового азота. Количество естественного (подчеркиваем) НБА в молоке по литературным данным составляет от 3−8% от всего азота, то есть средняя «ошибка» определения общего белка в молоке может составлять около 5%.
(NB!) Возможно, именно отсюда в наших нормативных документа (НД) возникает поправочный коэффициент, на который (согласно, например, стандартизированной методики ГОСТ 54 756–2011 — определение массовой доли сывороточных белков, поправка — 0,95) требуется умножить массовую долю определенного по Кьельдалю белка. Не совсем ясно к сожалению, почему именно на 0,95, ведь если мы определяем сывороточные белки должен учитываться НБА в сыворотке, а не в молоке.
В части собственно коэффициента имеются литературные данные, что первоначально данный коэффициент определялся не для смеси молочных белков, а для выделенного и условно чистого молочного казеина (sic !!!) и составлял 6,38. Он был получен из расчета, что в 100гр указанного белка содержалось 15,67 гр. азота. Долгое время это было общепринятое базовое значение, которое используется до сих пор. Понятное дело, что данный коэффициент имеет массу допущений — он никак не учитывает даже вариации макросостава белковых фракций молока, не говоря уже о генотипических различиях. Да и само значение (6,38) не является догмой во многих исследовательских работах для молока были предложены другие «уточняющие» коэффициенты, например, 6,34. Но на сегодня в большинстве случаев на официальном уровне (включая международную федерацию молока) продолжают использовать старый, и это трижды правильно! Почему, мы постараемся объяснить ниже.
Сначала про погрешности методов
Извечно возникающая в терминологии проблема, (весьма похожая на обсуждаемую далее проблему деления белка на истинный и общий), когда по факту нет четких и понятных разграничений в терминах и прописанных правил где и в каком случае эти термины применять. Для химика (не метролога) очевидно, что наиболее близок к истинному значению белок, напрямую выделенный и взвешенный, такие условно прямые методики измерения обычно стараются принять в качестве арбитражных (контрольных). У любых других косвенных методик анализа проблемы с пересчётом будут такие же, как и у методики Кьельдаля. Для биуратового метода будет аналогичный коэффициент, зависящий от количества пептидных связей, для его модификаций (Лоури) дополнительно еще и от количества тиоароматических групп. Понятно, что эти количества не являются константой и всегда будут зависеть от многих факторов, как и сам результат анализа соответственно. Отсюда согласно приятым в мире метрологическим «подходам» следует, что истинное значение может быть измерено, только с определённой долей неопределенности (не сочтите за каламбур), именно этот момент, то есть отсутствие абсолютно истинного значения будь то хоть трижды арбитражная методика или трижды государственный стандартный образец (СО), иногда трудно воспринимается россиянами, воспитанными исключительно в материалистическом духе. Также бывает проблематично понять и четко разделить распространяемые (если не сказать насаждаемыми) в текущем законодательстве типы неопределённостей (А и Б). Позволим себе упрощенно изложить наше частное видение термина неопределённости применительно к рассматриваемому случаю. Типа, А это неопределённость (кому удобно может считать это погрешностью) выведенная из формул математической статистики на основании некого конечного массива данных полученного экспериментально. Все формулы и правила их применения есть в учебниках, и именно эта неопределённость (типа А) как правило указывается в современных методиках (включая стандартизированные) и с большой долей вероятности приведенное в методике по определению белка в молоке методом Кьельдаля значение погрешности (0,03) получено именно таким способом. А вот неопределённость типа Б (В, англ.) это некая точностная характеристика которая учитывает ВСЕ наши знания об объекте и процессе проведении измерения, включая как объективные знания, так и субъективные. Эти знания можно упорядочить с помощью определённых математических приемов и включить, как это принято называть, в общий бюджет неопределённостей, но бывает это сделать проблематично или вообще невозможно. В былые времена в таких случаях использовался термин неисключённая систематическая погрешность, обычно она принималась равной нулю или пренебрежимо малой величиной, что иногда (не всегда к сожалению) оговаривалось в тексте самой методики.
Тут думается читателю было бы небезынтересно рассмотреть наши «подходы» при расчете неопределённости типа Б конкретно для случая определения общего белка в молоке по методике Кьельдаля. Ну самое первое — это учет погрешности пробоподготовки и средств измерений, участвующих в процессе анализа. Обычно для этого используют паспортные значения, например, погрешность (неопределённость) весов на которых мы взвешиваем навеску. Здесь в целом просто и понятно, например, если мы взвешиваем на аналитических весах с указанной паспортной погрешностью, то доля этой ошибки в общей неопредленности рассчитывается однозначно. Существуют также расчеты «ошибки эксперимента», например, имеются данные, где рассчитывались потери азота за счет образования комплекса с медью, если последняя использовалась в качестве катализатора при сжигании. Но бывает и более сложные случаи, в частности, как учитывать нелимитированные по сути выбросы и потери вещества (азота) при сжигании пробы, хотя конечно проще считать, что их нет, но по факту они существует. Точно также следует помнить, что в химии есть понятие «выхода реакции», и абсолютных (100%) выходов как правило не бывает, то есть во всех химических превращениях азота, прописанных в методиках использующих метод Кельдаля всегда существуют нерегламентируемые потери азота, аналогичные потери белка также существуют при разделении белковых фракций и в любых других процессах, переводящих белки из одного состояния в другое.
Вопросов здесь действительно много, но давайте вернемся к теме обсуждения. Если бы методика Кьельдаля подразумевала бы измерение аммонийного азота, то на этом можно было поставить точку и смысла считать далее неопределённость типа Б при пересчете азота на белок не было бы, но методика согласно своему названию подразумевает определение (измерение), а не расчет (термин — calculation в методиках ИСО) именно белка, поэтому мы обязаны включить в бюджет неопределенности любые известные нам ошибки коэффициента. Давайте попробуем сделать это применив как указано выше ВСЕ наши знания о предмете.
Итак, если читатель помнит, то коэффициент пересчета был изначально (более века назад) рассчитан и предложен для кислотно осажденного и очищенного казеина Хамарстеном и Сибелиенем (приносим извинения за вольный перевод фамилий), но в последствии были и другие расчеты, для молока предлагались значения 6,34, 6,30 и даже общепринятый для большинства пищевых объектов 6,25. В целом анализ имеющихся данных позволяет утверждать, что суммарный коэффициент для казеиновой фракции отличен как от альбуминовой, так и от фракции небелкового азота и в целом варьируется в интервале от 5,8 до 6,6 для индивидуальных и групповых белков молока. Более того коэффициент для одних и тех же молочных белков (например, альбумина), но полученный из разных источников тоже может варьироваться (до 0,1ед), это связано с точностью измерения собственно количества азота которая зависит от точности определения массы пробы при сжигании, а также вероятных вариаций состава в зависимости от источника получения образца и способа его очистки.
Далее следует рассмотреть к какому объекту мы в реальности применяем обсуждаемый коэффициент и какой белок рассчитываем. Здесь следует понимать, что молоко — это не искусственная смесь очищенных белков, белковая система молока состоит из весьма сложных белковых образований, отличающихся между собой как по составу, так и по своей пространственной структуре и взаимодействию с другими небелковыми элементами системы. Кроме этого существуют еще дополнительные условно белковые соединения (например, протеиды имеющие в составе неорганическую или липидную часть). Считать или не считать такие соединения белками и включать или не включать их в расчет общего белка на сегодня не определено. Аналогично можно отметить, что даже исключительно молочный белок — казеин тоже существует в виде мицелл сложного состава. Например, примерно 20% кальция, содержащегося в молоке, находится в виде комплекса с казеином. Будет ли данный кальций частью белковой фракции, а если да нужно ли тогда вычитать его массу из массы минеральных солей определённых при сжигании.
Также следует учитывать условия пробоподготовки и, обработки и собственно объект исследования. Так, например, очевидно, что при анализе по Кьелдалю казеина (на фильтре после осаждения) и сывороточных белков (в фильтрате) следует применять кардинально различные коэффициенты.
Как видим вопросов больше чем ответов, но в целом суммируя сказанное выше лично мы делаем (подчеркиваем мы, то есть это только наше мнение) вывод, весьма простой по логике, но нетривиальный с точки зрения существующего законодательства, что коэффициент может и должен быть разным с учетом всех известных случайных и систематических ошибок и подбирается индивидуально для конкретной лаборатории и объекта анализа. И в подтверждение сказанному, мы в явном виде указываем, какие именно коэффициенты пересчета применяются в лаборатории ООО НПП «БИОМЕР»:
— для молока 6,38,
— для фракции сывороточных белков 6,28
— для казеиновой фракции 6,45.
и именно эти значения будут указаны в любом нашем протоколе анализа, пока лаборатория по каким-либо причинам не примет решение их изменить.
Откуда взялись эти цифры.
— во-первых, следует помнить, что метод Кьельдаля — это только один из методов, и имеются другие способы определения белка, в которых не существует ошибки связанной с НБА.
— во-вторых существует определённые химические приемы по обработке пробы, которые позволяют исключить влияние НБА на конечный результат.
— ну и в-третьих всегда есть дополнительные косвенные данные позволяющие подтвердить или опровергнуть результат, полученный по методу Кьельдаля.
По последнему пункту отдельно хотелось бы уделить внимание «материальному балансу» при анализе молока. В кавычках потому что он конечно весьма условен и существует в рамках определённых допущений и погрешностей измерения каждого компонента. Поясним. Теоретически кто бы что и как не толковал современные способы подсчёта общего весового состава молока общепринято, что сухие вещества молока состоят из жировой, углеводной, белковой и минеральной части.
То есть сухой молочный остаток (СМО)=Жир+Белок+Лактоза+Соли.
Это подчеркиваем некое базовой понятие, можно далее рассуждать, что не весь жир — триглицериды, не все углеводы лактоза, а уж органические кислоты или мочевину отнести к солям, так вообще невозможно ни по названию, ни по способу измерения этих самых минеральных солей. Повторимся это допущения (как и те что описывались выше, про кальций итп), можно ли ими пренебречь при расчёте баланса или нужно обязательно учитывать решается индивидуально для конкретного случая и лаборатории, но в целом подчеркиваем, это должно соблюдаться поскольку это общая балансная база. Лично мы ее соблюдаем. Баланс должен «биться» (хим.), для нас СОМО это всегда СМО- Жир, и одновременно Лактоза+Белок+Соли (зола), а не Лактоза+Белок+Соли+некая известная\или неизвестная сущность.
Более того, существуют еще другие базовые измеряемые макропоказатели молока, в частности действительная плотность, одна тоже входит в баланс поскольку «общая» плотность складывается из плотностей молочных составляющих, или точка замерзания, которая зависит практически исключительно от количества растворенных веществ в плазме молока. Сообразно сказанному даже теоретически плотность молока не может выходить из регламентированного составом диапазона, и точка замерзания должна зависеть в первую очередь от массовой доли лактозы, а не титруемой кислотности или соматических клеток. Если это не так, значит в измерениях существует аналитическая ошибка, либо объект — анормальное молоко, либо вообще не молоко. Все сказанное можно отнести и к балансу общего белка, то есть.
Общий Белок = Казеин + Сывороточные Белки.
Таким образом суммируя и памятуя ВСЕ сказанное выше мы стараемся использовать систематически обоснованный коэффициент, и любые полученные нами данные единичного измерения, должны (по возможности конечно) быть подкреплены дополнительными анализами и расчетами. И именно из систематических данных которые были собраны и проанализированы на протяжении не одного года появились указанные выше коэффициенты, и именно эти значения (6,38, 6,28, 6,45) используются при калибровке наших приборов.
Но мы ни коим образом не считаем их догмой и, если мы обнаружим какой-либо фактор (неопределённость типа Б) их уточняющий мы обязательно будем его использовать и учитывать.
Политическо-законодательные аспекты.
Для этого сразу следует задаться вопросом, а почему хотя проблема коэффициента пересчёта и НБА известна давно химики аналитики на практике не жалуются о систематическом завышении содержания белка по сравнению с другими методиками (а это не много ни мало примерно 0.19%) и тем более не спешат решить проблему кардинально, то есть не используют никаких поправочных коэффициентов и продолжают рассчитывать белок по базовому коэффициенту (6,25 или 6,38), хотя можно было бы для молока сразу взять к примеру, 6,06, учтя таким образом среднее значение НБА. А происходит это (по нашему мнению, конечно) по той простой причине, что введение поправочных коэффициентов или что еще хуже перманентная актуализация коэффициентов пересчёта нарушает саму методологию анализа, анализ по Кьельдалю предназначен для определения содержания общего азота, и расчета (не измерения) общего (crude) белка. Для определения любого истинного (true) белка, будь то общая масса или масса отдельных фракций, существуют другие методики и способы, и анализ азота по Кьельдалю (или Дюма) при этом может быть частью такого способа. Введение любого волшебного поправочного коэффициента не превратит автоматически общий белок в истинный. Методологически в таком подходе мы тогда получаем даже не измеренное значение общего азота, а некое расчетное значение белка, которое будет возможно разумным, но к реальному измерению собственно белка никакого отношения не имеющее.
А как у них?
А у них в целом нет такой проблемы, просто в разных случаях используются разные «базы и уровни». На международном уровне в качестве базового значения для расчета за товар (payment testing) и показателей продуктов питания используется значения общего белка. Некоторые страны у себя используют для этих же целей и истинный белок. А вот для исследовательских целей вполне логично использовать значения истинного белка, что и делается. Вообщем все индивидуально.
Ну и напомним, что существует ИСО 8968, состоящих из N частей, в каждой из которых подробно описаны и сам метод, и коэффициенты, и полученные рассчитанные значения, и смысл, того, зачем и как это делается. Каким образом из «Определения азота по Кьельдалю и расчета содержания общего белка» у нас получается «Определение массовой доли белка методом Кьельдаля» вопрос риторический, но может в этом и есть корень зла?
Что будет?
Ну предположим на законодательном уровне для молока будет принято решение что-то изменить, тогда в первую очередь следует понимать проблемы, которые при этом возникнут.
- При переходе на истинный белок (например, путем применения зачастую используемой аддитивной поправки 0,19) все полученные результаты анализа белка по Кьельдалю станет невозможно сравнивать со старыми цифрами, прописанным в нормативно-правовых актах и архивных данных, включая статистические.
- Каким образом будет регламентировано нахождение соответствия между полученным таким образом истинным белком и белком, полученным по другим методиками и в частности инструментальными методами с использованием средств измерений, отградуированных или поверяемых по тому же методу Кьельдаля. Особенно в случае, если отдельно не выделено и не оговорено какой белок общий\истинный измеряется прибором.
- В каком документе будет прописано и регламентировано применение поправочного коэффициента для расчета истинного белка. Как этот документ будет соотносится с уже имеющимися.
- Кто (какая конкретно организация) и на основании каких данных (эксперимент, литературные данные) в итоге будет определять конкретное значение коэффициента.
- Как (технически) и в какие сроки будет осуществляться переход при внедрении термина истинного белка для расчета за товар. Как при этом будет урегулированные вопросы ценовой конкуренции.
Сразу оговоримся в случае принятия решения о неком принципиальном методологическом переходе с общего на истинный белок мы не призываем к его саботажу и не генерируем проблемы, и в целом не видим принципиальной разницы какой белок истинный или общий берется в качестве результата, мы за то, чтобы все пересчеты делались осмысленно и взвешено. Использование надуманного поправочного коэффициента и его административное введение особенно в рамках одного объекта анализа (молочка) и государства (РФ), окончательно запутает и терминологию и данные лабораторий и взаиморасчёт за товар. В последней части (мы уже писали об этом здесь) правильно, по нашему мнению, будет в случае использования методики анализа белка по Кьельдалю сразу фиксировать на уровне договоров коэффициент белка, который будет использоваться при расчете за товар. А лаборатория соответственно должна указывать в протоколе или в явном виде коэффициент пересчёта или только полученные значения по содержанию азота, оставляя способы пересчёта и абсолютное значения коэффициентов на усмотрение продавца и покупателя.
Ну вот все что хотелось бы сказать по данной теме на сегодня.
Спасибо за терпение тем, кто прочел до конца, и за любые комментарии по делу.
Необходимость определения содержания белка в зерновых культурах в нашей стране уже ни у кого не вызывает сомнений. Возникает вопрос: как это лучше сделать? По нашему мнению, содержание белка нужно определять только классическим методом.
Датский химик Иоганн Кьельдаль разработал метод анализа содержания азота в органических веществах, известный в настоящее время, как метод Кьельдаля.
Метод включает в себя три основных этапа: дигерирование, дистилляцию и титрование. В данной статье рассматривается наиболее важный и сложный этап — исходная процедура дигерирования.
Для упрощения процедуры анализа по Кьельдалю компанией Foss Tecator была разработана простая и надежная методика дигерирования, которую можно использовать практически для любых образцов. Эта базовая процедура позволяет анализировать более 90 % протеиносодержащих и более 60 % всех других азотосодержащих образцов. Вкратце процедуру дигерирования можно описать следующим образом:
Необходимое условие получения точных результатов анализа по Кьельдалю — тщательная подготовка образцов. Процедура подготовки проб проводится в один или несколько этапов и должна обеспечивать гомогенизацию образца, т. к. размер частиц в анализируемых пробах не должен превышать 1 мм. Однородность образца повышает воспроизводимость метода, а также позволяет уменьшить объём пробы. Обычно при использовании частиц малых размеров повышается скорость дигерирования. Поэтому для подготовки образцов необходима хорошая лабораторная мельница.
Взвешивание образцов для последующего анализа по Кьельдалю должно проводиться на аналитических весах с точностью до 0,1 мг.
Ориентировочный объём образца можно определить, руководствуясь следующими рекомендациями:
| Содержание протеина | Вес образца, мг |
| До 5 % | 1000 – 5000 |
| 5 – 30% | 500 — 1500 |
| Более 30 % | 20 – 1000 |
Важно знать влажность образца и всегда анализировать только предварительно высушенные образцы. Если влажность образца известна, то для вычисления содержания протеина в сухом образце можно воспользоваться такой формулой:
Р = А * 100 / (100 — В),
где А — измеренное содержание протеина в образце, (%)
В — содержание влаги в образце, (%).
Цель процедуры дигерирования — полное разрушение в образце химических связей азота и преобразование всего азота в ионы аммиака. До настоящего времени не найдена замена серной кислоте, которая используется для вышеуказанной цели. Однако использовать для дигерирования чистую серную кислоту нецелесообразно из-за низкой скорости протекания процесса. Скорость дигерирования и разрушения образца зависят не только от свойств кислоты, но и от температуры обработки. Чем выше температура, тем меньше времени уходит на дигерирование. При использовании чистой серной кислоты температура дигерирования ограничивается, в основном, её точкой кипения (338 оС). Необходимо отметить, что критическая температура, необходимая для разложения, также высока и равна 373 оС. Скорость дигерирования, можно значительно увеличить добавлением соли и катализаторов.
В классическом аналитическом приборе Кьельдаля на каждый грамм образца обычно необходимо 25 мл кислоты, в то время как в блочных дигесторах компании Foss Tecator благодаря оптимизации процесса на грамм вещества, как правило, требуется лишь 12 мл кислоты.
Объём серной кислоты, используемый для анализа, можно оптимизировать с учетом конкретного типа образца. Следует учитывать количество кислоты, которое будет потеряно на испарение, а также вступит в реакцию с другими реактивами и с образцом. Другой немаловажный фактор — потребление кислоты. При увеличении количества кислоты на стадии дигерирования необходимо пропорционально увеличить количество щелочи на этапе дистилляции.
Допустим, исследуемый образец пшеницы имеет такой состав:
Протеин 12,5 %
Углеводы 66,5 %
Жир 3,5 %
Для дигерирования 1 г такого образца потребуется такое количество кислоты:
Протеин 12,5% * 1,0 * 4,9 = 0,61 мл
Углеводы 66,5% * 1,0 * 4,0 = 2,66 мл
Жир 3,5% * 1,0 * 4,7 = 0,34 мл
Всего 3,61 мл
Замена классической системы дигерирования по Кьельдалю блочным дигестором с эффективной вытяжной системой позволяет минимизировать выделение в окружающую среду большей части газообразных продуктов реакции.
В табл. 1 приведен краткий обзор общего потребления кислоты при дигерировании.
| Таблица 1. равнение потребления кислоты при дигерировании с использованием классической системы Кьельдаля и блочного дигестора компании Foss Tecator | ||
| Блочный дигестор | Классическая система Кьельдаля | |
| Используемый объём кислоты | 12 мл | 25 мл |
| Потери при испарении | 1,2 мл | 7,2 мл |
| Потребление на 1 г образца | 3,6-7 мл | 3,6-7 мл |
| Потребление реактивами | 2,1 мл | 4,2 мл |
| Остаток в пробирке для дигерирования | 1,7-5,1 мл | 6,6-10,0 мл |
| Используемый объём щёлочи | 50 мл | 100 мл |
Таблица наглядно иллюстрирует два основных преимущества блочного дигестора (если рассматривать экологический аспект): минимизацию потерь кислоты при испарении и сокращение объёма гидроксида натрия, добавляемого на этапе дистилляции.
Время дигерирования можно уменьшить путем увеличения температуры кипения серной кислоты, выбирая оптимальное соотношение кислота/соль. Типичное исходное соотношение кислота/соль лежит в диапазоне от 1,4 до 2,0.
Лучший способ избежать вариаций при добавлении соли — использовать стандартизированные изделия, например, таблетки Кьельтабз.
Скорость и эффективность дигерирования зависят не только от температуры. Добавление соответствующего катализатора также помогает оптимизировать эти параметры. Сегодня всё большее распространение получают катализаторы на основе меди и, до некоторой степени, селена.
Уменьшить время дигерирования также возможно при помощи введения в реакционную смесь окислителей. В современных методиках дигерирования в качестве окислителей обычно используют персульфат калия или перекись водорода.
Из этих двух веществ более широко используется перекись водорода, выполняющая две основные функции:
1.Является очистителем, ускоряет разложение органического материала.
2.Подавляет пенообразование, регулируя выделение пены в ходе дигерирования.
Добавлять перекись водорода рекомендуется в малых количествах (не более 5 мл) так, чтобы она медленно стекала по внутренней поверхности пробирки для дигерирования.
Полное время, необходимое для дигерирования, как отмечалось выше, зависит от многих факторов: типа образца, объёма кислоты, количества соли, катализатора, окислителя, реагента, используемого для разложения, восстанавливающего реагента, температуры блочного дигестора.
Существует также понятие «время кипячения», которое включает в себя два этапа обработки образца.
На первом этапе дигестат осветляется или становится бесцветным. Продолжительность этого этапа обычно называют «временем дигерирования». На втором этапе оставшийся азот преобразуется в кипящей кислоте в форму, доступную для дистилляции. Это — так называемый «период кипячения».
Легко проследить ход дигерирования, регулярно прерывая процесс и определяя содержание азота в дигестате. Процесс восстановления азота при дигерировании можно представить на графике (рис. 1).

График показывает, что образцы различных типов ведут себя по-разному. Для одних полное дигерирование достигается в точке осветления (вещество В), другие нуждаются в длительном периоде дигерирования (вещество А). Для вещества А процесс восстановления азота идет медленно, время дигерирования сравнительно велико и для 99-процентного восстановления азота полная продолжительность обработки образца должна составить около 60 мин. В точке осветления обычно достигается восстановление до 95-100% азота.
Одной из проблем, которые могут возникнуть при анализе по методу Кьельдаля, является пенообразование, возникающее иногда на начальной стадии дигерирования.
Эта проблема решается двумя способами:
— используются стеклянные стерженьки для кипячения;
— добавляется перекись водорода.
Если пенообразование — единственная возникающая проблема, то лучше добавить 1-3 капли октанола или соответствующую подавляющую пенообразование эмульсию.
Итак, нами рассмотрен ряд факторов, влияющих на определение содержания азота в образцах как с помощью классического, так и усовершенствованного метода. Усовершенствованный метод компании Foss Tecator позволяет повысить скорость и точность процедуры дигерирования, уменьшить габариты оборудования, сократить потребление энергии и химических реагентов.
В табл. 2 классическая процедура дигерирования по Кьельдалю сравнивается с работой блочного дигестора компании Foss Tecator.
Как видно из табл. 2, использование блочной системы Foss Tecator сокращает общее время дигерирования в три раза, потребляемую мощность на 95 %, а также на 50 % снижает количество реагентов, необходимых для анализа. Всё это существенно улучшает параметры классической процедуры.
Ковтонюк Е. Н., генеральный директор НА «Укрзернопродукт»
Острик Н. М., кандидат сельскохозяйственных наук
Метод Кьельдаля, определение азота и белка
Разбираем сущность метода Кьельдаля. Как проводить разложение, дистилляцию и титрование. Определение содержания азота и белка.
Метод Кьельдаля, определение азота и белка
Разбираем сущность метода Кьельдаля. Как проводить разложение, дистилляцию и титрование. Определение содержания азота и белка.
Метод определения азота по Кьельдалю был разработан в 1883 году в химической лаборатории датской пивоваренной компании Карлсберг, где Йохан Кьельдаль занимался исследованием процессов брожения. Он выявил зависимость готовности пива от содержания белка в сусле и охарактеризовал её так: «Less protein meant more beer» Меньше белка – больше пива.
Азот являются строительным материалом для любых клеток, и он также важен для роста и размножения клеток дрожжей.
Сущность метода Кьельдаля заключается в переводе органического азота в аммонийную форму, в процессе термического разложения образца с кислотой, дальнейшая дистилляция полученной смеси со щелочью для перевода аммонийного азота в аммиак, поглощение аммиака водным раствором и титрование полученного раствора. Метод Кьельдаля является универсальным и используется для определения содержания азота в пищевых и фармацевтических продуктах, текстиле, полимерах, объектах окружающей среды и т.д.

Следует различать понятия общий азот по Кьельдалю и общий азот. Общий азот по Кьельдалю обозначается TKN (Total Kjeldahl Nitrogen) и представляет собой сумму органических форм азота и аммонийного азота. При анализе почв и сточных вод помимо органического и аммонийного азота также будет учитываться аммиачный азот.
Азот содержащейся в виде нитратов и нитритов при анализе по методу Кьельдаля не учитывается. Общее содержание азота во всех органических и неорганических формах обозначается TN (Total Nitrogen) и определяется как сумма азота по Кьельдалю и азота в форме нитратов и нитритов.
Органический азот гетероциклических соединений при разложении по Кьельдалю не переходит в аммонийную форму, поэтому образцы с гетероциклами азота не исследуют методом Кьельдаля.
Общий азот по Кьельдалю

В исследовании Кьельдаля, источником азота для дрожжей служили белки пивного солода (злаков). Для определения содержания азота, Кьельдаль создал установку разложения органических продуктов и сформулировал метод количественного определения азота. Спустя несколько лет, методика определения азота по Кьельдалю была опубликована в научных журналах Германии и Франции, и получила широкое распространение. В наши дни метод определения азота по Кьельдалю является референтным методом и используется для проверки правильности определения азота другими методами.
Разложение по Кьельдалю
Процедура разложения пробы по методу Кьельдаля является самой продолжительной и трудоемкой стадией анализа. На этой стадии происходит перевод органических форм азота в аммонийный азот, процесс протекает при нагревании под действием серной кислоты в присутствии катализаторов (сульфата меди, сульфата ртути). В классическом методе разложение проводят в круглодонных колбах помещенных в песчаную баню или колбонагреватель. Кьельдаль использовал для разложения круглодонные колбы с длинным горлом, позже их стали именовать колбами Кьельдаля.
Скорость и полнота разложения пробы зависят от температуры, чем выше температура, тем эффективнее протекает разрушение матрицы и образование сульфата аммония. При этом чрезмерный нагрев образца может привести к потере азота, а занижение температуры к неполному извлечению азота из матрицы. Ошибки при выполнении разложения по Кьельдалю могут быть критичны для достоверности и повторяемости результата анализа. Именно в проведении стадии разложения содержаться основные отличия анализа по Кьельдалю различных образцов.
Контроль процесса разложения по Кьельдалю
В ходе разложения органических образцов происходит вспенивание и обугливание органической матрицы, смесь в сосуде приобретает черный цвет. Об окончании процесса разложения свидетельствует образование в реакционной колбе прозрачного раствора.
Полноту извлечения и отсутствие потерь в процессе разложения по Кьельдалю контролируют с помощью стандартных веществ с известным содержанием азота, которые вносят в одну из реакционных колб. Для легкоразлагаемых проб используют N-фенилацетамид C8H9ON, содержащий 10,36 % азота. Для трудно минерализуемых проб используют триптофан C11H12N2O2 , содержащий 13,72% азота. Извлечение азота для N-фенилацетамид должно быть не менее 99,5%, для триптофана не менее 99%.
Для упрощения этого трудоемкого процесса, минимизации потерь и получения достоверных результатов многие производители лабораторного оборудования разрабатывают системы разложения (дегисторы). Дегистор создает повторяемые условия разложения нескольких образцов и утилизирует едкие пары кислот.
На российском рынке представлены дегисторы для разложения по Кьельдалю на 4, 6, 8, 12 и 20 образцов отечественных (ЛОиП, Вилитек) и импортных производителей (Velp, Gerhardt, Buchi).

CHNO + H2SO4→ H2O + CO2 + SO2 + (NH4)2SO4
Дистилляция (перегонка с паром)
На этой стадии переводят аммонийный азот в аммиак и отгоняют полученный аммиак с водяным паром. Аммиак выделяется в ходе реакции смеси полученной на стадии разложения с концентрированной щелочью. Раствор после стадии разложения содержит избыток серной кислоты, поэтому щелочь берут в избытке, для нейтрализации серной кислоты и реакции сульфатом аммония.
В классическом варианте содержимое колбы Кьельдаля количественно переносят в перегонную колбу, добавляют дистиллированную воду и избыток концентрированного раствора гидроксида натрия. Перегонную колбу нагревают и перегоняют аммиак в приемную колбу. Скорость перегонки зависит от температуры, однако, даже при слабом нагреве весь аммиак отгоняется в течение 10 минут. Для поглощения паров аммиака в приемную колбу добавляют 3-4 % борную кислоту, либо стандартизованный раствор соляной кислоты.
Чтобы сократить ручные операции и уменьшить потерю аммиака производители лабораторного оборудования предлагают дистилляторы для автоматизации этой стадии. В дегисторах и дистилляторах Кьельдаля одного производителя сосуды для разложения пробы совместимы с установками для дистилляции, это позволяет избежать количественного переноса вещества из одного сосуда в другой. Дозирование раствора щелочи, разбавление образца и подача пара со встроенного парогенератора в таких приборах автоматизированы и внесены во встроенные программы.
Качество процесса перегонки с паром можно проконтролировать добавлением известного количества сульфата аммония или хлорида аммония в сосуд перед началом процесса отгонки. Степень отгонки аммиака в приемный сосуд должна быть не менее 99,5%. В противном случае необходимо выбрать другую программу перегонки.

(NH4)2SO4 + 2NaOH → 2NH3 + Na2SO4 + 2H2O
Поглощение аммиака раствором соляной кислоты
2NH3 + 4H3BO3 → (NH4)2B4O7 + 5H2O
Поглощение аммиака раствором борной кислоты
Титрование по Кьельдалю
Количество аммиака в приемной колбе определяют титрованием.
Если в качестве поглотителя в приемной колбе использовалась сильная кислота (HCl, H2SO4), то титрование проводят раствором щелочи. Это будет обратное титрование, в ходе него титруют избыток сильной кислоты, оставшейся после реакции с аммиаком. Точку эквивалентности определяют потенциометрически или при помощи индикатора метиловый красный.
Если поглотителем в приемной колбе был раствор борной кислоты, то в качестве титранта используют соляную кислоту. Титрование проводят вручную или с использованием титратора, определяя точку эквивалентности потенциометрически или с использованием смешенного индикатора метиловый красный + метиленовый голубой. Поглотительный раствор с борной кислотой необходимо титровать сразу после окончания процесса дистилляции, чтобы избежать потери аммиака.
Изначально Кьельдаль использовал в качестве поглотителя аммиака раствор соляной кислоты, и содержание аммиака определял йодометрическим титрованием. В наше время большее распространение получило использование в качестве поглотителя 4% борной кислоты и титрование раствором щелочи.
На сегодняшний день автоматизирована и стадия титрования. Современные производители анализаторов Кьельдаля совмещают титраторы с дистилляторами. Датчики титратора располагаются в приемном сосуде дистиллятора, и титрование проводится автоматически по завершению процесса отгонки с паром.

После завершения титрования рассчитывают массу азота в анализируемом образце.
mN = ((Vхол – Vобр) * СNaOH * TITER * 14) / 1000
mN = ((Vобр – Vхол) * СHCl * TITER * 14) / 1000
Расчет белка по методу Кьельдаля
Органически связанный азот содержится в белках, аминокислотах, нуклеиновых кислотах и играет важную роль в метаболизме живых организмов. Содержание азота в различных индивидуальных молекулах белка колеблется в диапазоне от 14 до 19%. Обычно при пересчете содержания азота в содержание белка используется усредненное содержание азота в белке – 16 %. Таким образом, коэффициент пересчета на белок равен 100/16=6,25.
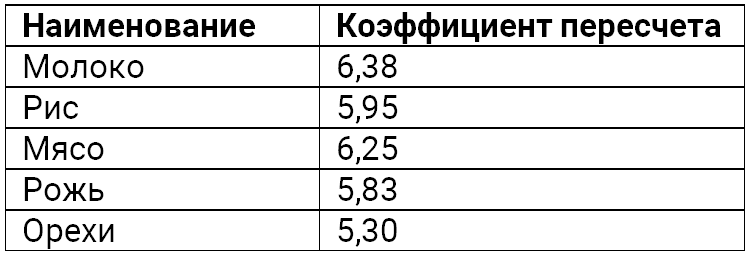
В некоторых продуктах питания преобладают белки определенного типа, для них определены другие коэффициенты пересчета.
Методики определения азота по Кьельдалю
ГОСТ 23327-98, Молоко и молочные продукты. Метод измерения массовой доли общего азота по Кьельдалю и определение массовой доли белка
ГОСТ 26889-86, Продукты пищевые и вкусовые. Общие указания по определению содержания азота методом Кьельдаля
ГОСТ 28743-93 (ИСО 333-96), Топливо твердое минеральное. Методы определения азота
ГОСТ 34111-2017, Продукция соковая. Определение содержания азота методом Кьельдаля
ГОСТ 34454-2018, Продукция молочная. Определение массовой доли белка методом Кьельдаля
ГОСТ 34536-2019, Молоко и молочная продукция. Определение массовой доли сывороточных белков методом Кьельдаля
ГОСТ 34789-2021, Продукция пивоваренная. Идентификация. Определение массовой концентрации общего азота методом Кьельдаля
ГОСТ 17837-2013, Продукты сырные плавленые. Определение содержания азота и расчет содержания общего белка. Метод Кьельдаля
ГОСТ Р 54662-2011, Сыры и сыры плавленые. Определение массовой доли белка методом Кьельдаля
ГОСТ Р 55246-2012, Молоко и молочные продукты. Определение содержания небелкового азота с применением метода Кьельдаля
ОФС.1.2.3.0011.15, Определение азота в органических соединениях методом Кьельдаля
ПНД Ф 14.1:2:4.277-2013 Количественный химический анализ вод. Методика измерений массовых концентраций азота органического методом Кьельдаля в питьевых, природных и сточных водах
ПНД Ф 16.1:2:2.3.82-2013 Количественный химический анализ почв и отходов. Методика определения азота общего методом Кьельдаля в осадках сточных вод, органических удобрений, грунтах тепличных и почвах
ASTM D3228-22, Standard Test Method for Total Nitrogen in Lubricating Oils and Fuel Oils by Modified Kjeldahl Method
ASTM D3590-17, Standard Test Methods for Total Kjeldahl Nitrogen in Water
Вам также может быть интересно:
-
Введение
-
Разложение пробы
-
Дистилляция
-
Титрование
-
Расчет белка
-
Методики